Small Molecule Drugs vs Gene Replacement: A Comparison
Small molecules give accessible symptom control; gene replacement targets genetic causes but brings higher cost and delivery risks.
When it comes to treating rare genetic diseases, two approaches dominate: small molecule drugs and gene replacement therapies. Both have unique methods, benefits, and challenges. Here’s what you need to know:
- Small Molecule Drugs: These are typically pills or injections that adjust cellular pathways or stabilize proteins. They require regular dosing but are often more accessible and cost-effective.
- Gene Replacement Therapy: This one-time treatment introduces a functional gene to address the root cause of a condition. It offers long-term effects but comes with high costs and delivery challenges.
Quick Comparison
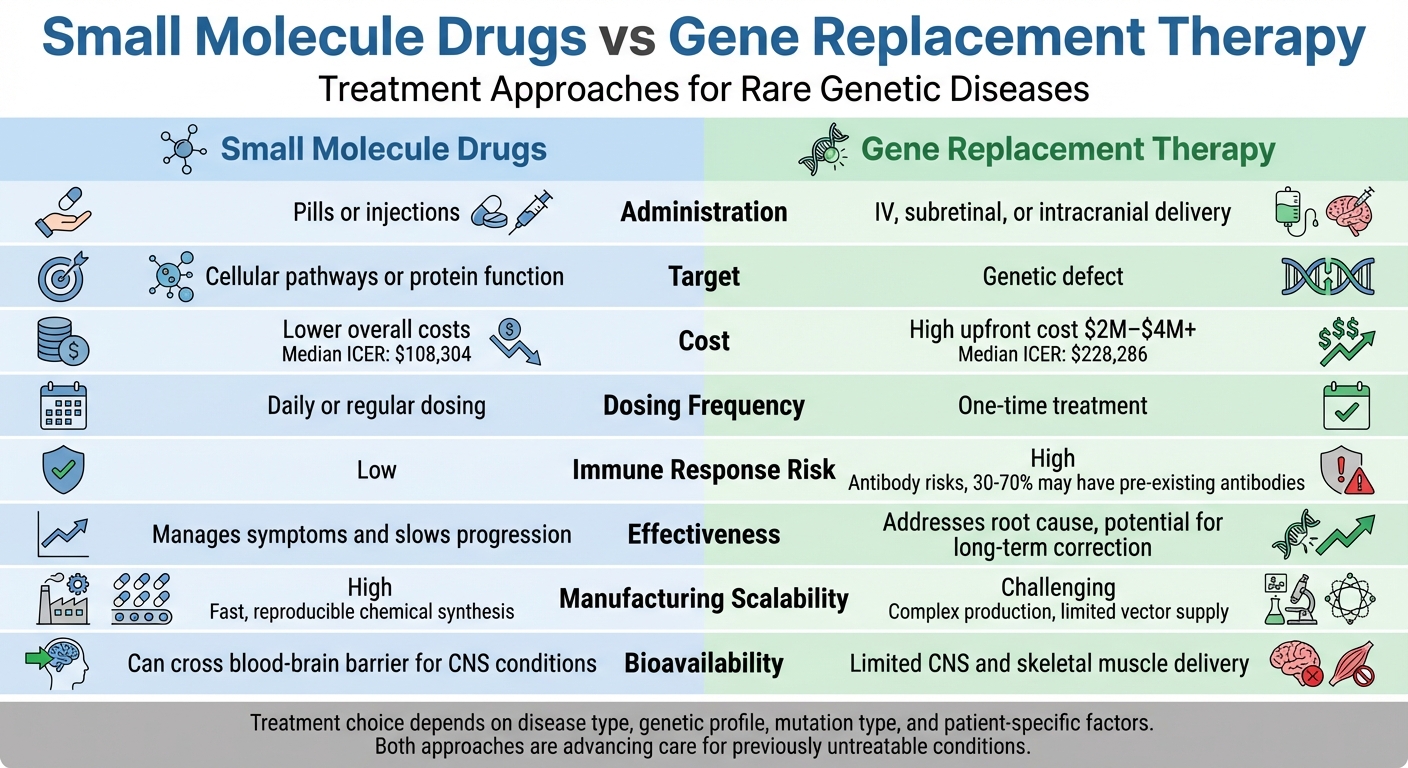
| Feature | Small Molecule Drugs | Gene Replacement Therapy |
|---|---|---|
| Administration | Pills or injections | IV, subretinal, or intracranial delivery |
| Target | Cellular pathways or protein function | Genetic defect |
| Cost | Lower overall costs | High upfront cost ($2M–$4M+) |
| Dosing Frequency | Daily or regular | One-time treatment |
| Immune Response Risk | Low | High (antibody risks) |
| Effectiveness | Manages symptoms | Addresses root cause |
Choosing the right treatment depends on factors like the condition, patient’s genetic profile, and accessibility. Both approaches are advancing rare disease care, offering hope for conditions that were previously untreatable.
Small Molecule Drugs vs Gene Replacement Therapy Comparison Chart
How These Therapies Work
Small Molecule Drugs: Modifying Cellular Pathways
Small molecule drugs are tiny compounds, typically weighing under 900 Daltons, that can easily slip through cell membranes. Their goal? To tweak enzymes, proteins, or receptors - essentially fine-tuning cellular pathways rather than replacing what's broken.
These drugs operate in various ways: they can stabilize proteins (like Acoramidis, approved in 2024 for ATTR‐Cardiomyopathy, which stabilizes the transthyretin protein), inhibit enzymes to prevent harmful substance buildup, or even enhance pseudoexon inclusion to reduce toxic protein levels. A promising example is an oral brain-penetrant candidate showing potential in Huntington's disease.
"Small molecule drugs have the advantage of easily penetrating cell membranes and affecting intercellular targets such as enzymes and receptors that biologics cannot reach."
– Phil McHale, Consultant and Cheminformatics Expert
However, these drugs come with limitations. They often manage symptoms and slow disease progression rather than fixing the underlying genetic problem. Regular dosing, like daily pills or injections, is usually required because their effects fade as the drug is cleared from the body. Despite this, their ability to cross the blood-brain barrier makes them particularly valuable for central nervous system conditions. For example, Trofinetide (DAYBUE), approved in 2023 for Rett Syndrome, has demonstrated meaningful neurological improvements.
While small molecule drugs focus on chemical adjustments, gene replacement therapy takes a more direct approach by addressing the genetic root of the disorder.
Gene Replacement Therapy: Inserting Working Genes
Gene replacement therapy involves delivering a functional copy of a defective gene into patient cells, often using modified adeno-associated viruses (AAVs) as carriers.
"Think of vectors like envelopes being used to deliver a specific message. The envelope allows the message to be delivered, while the message inside provides new instructions to the cell on how to function properly."
– ASGCT
There are two main delivery methods. In vivo delivery injects the vector directly into the body to target specific organs like the liver, eyes, or nervous system. Ex vivo delivery, on the other hand, involves removing patient cells, modifying them in a lab, and reintroducing them. A great example of this is Zevaskyn (pz-cel), approved in 2025 for Recessive Dystrophic Epidermolysis Bullosa.
Once the vector reaches its destination, it enters the cell and delivers genetic instructions to the nucleus. Non-integrating vectors, such as AAVs, keep the new DNA separate from the host genome, making them ideal for non-dividing cells like those in the brain or heart. Meanwhile, integrating vectors like lentiviruses embed the genetic material directly into chromosomes, ensuring the new gene is copied during cell division. Upstaza (eladocagene exuparvovec), approved in November 2024 for AADC deficiency, was the first gene therapy delivered directly to the brain through a surgical procedure.
Gene replacement therapy is designed as a one-time treatment with long-lasting effects. Still, challenges remain. Between 30% and 70% of patients may already have antibodies against AAV vectors, potentially neutralizing the therapy. Additionally, the liver often captures over 90% of systemically infused AAV vectors, limiting their availability to other organs. These hurdles underscore the complexity of delivering such treatments effectively.
Main Differences in How They Work
The key difference between small molecule drugs and gene replacement therapy lies in their approach to cellular dysfunction. Small molecule drugs tweak existing cellular pathways and protein functions, acting like precision tools. Gene replacement therapy, however, tackles the root cause by providing a working gene, enabling cells to produce the correct protein themselves.
The trade-off is clear: small molecule drugs offer convenience and accessibility through ongoing dosing, while gene replacement therapy aims for a one-time, long-term solution by directly addressing the genetic defect.
What is Gene and Cell Therapy - 3D Animation | Mass General Brigham
FDA-Approved Examples in Rare Genetic Diseases
Several FDA-approved treatments showcase how these therapies are applied to tackle rare genetic diseases effectively.
Small Molecule Drug Examples
Migalastat (Galafold), approved in 2018 for Fabry disease, works by stabilizing the misfolded α‐galactosidase enzyme, allowing it to regain lysosomal activity. Clinical trials have shown a noticeable reduction in cardiac mass, with 35–50% of patients qualifying for treatment after genetic testing.
Eliglustat (Cerdelga), approved in 2014 for Gaucher disease, targets the buildup of toxic glucosylceramide by inhibiting specific enzymes. This treatment is conveniently administered orally.
Risdiplam (Evrysdi), approved in 2020, enhances the production of functional SMN protein by modifying SMN2 splicing. Meanwhile, Tafamidis (Vyndaqel), approved in 2019, stabilizes transthyretin to stop the formation of amyloid fibrils.
Gene Replacement Therapy Examples
Zolgensma uses a single intravenous infusion to deliver a functional SMN1 gene. In Phase 1 trials, 100% of infants with SMA Type 1 survived without permanent ventilation, compared to just 8% historically. However, the treatment comes with a hefty price tag of $2.10 million per dose.
Luxturna addresses RPE65-related retinal dystrophy through a direct injection into the retina. Clinical trials revealed that 62% of treated patients passed a mobility test at 1 lux lighting conditions, while none in the control group did.
Hemgenix, approved for hemophilia B, introduces a functional Factor IX gene to the liver, with a cost of $3.50 million per dose. Similarly, Lenmeldy, set for approval on March 18, 2024, targets metachromatic leukodystrophy and is priced at $4.25 million, reflecting the growing trend of one-time, gene-specific treatments.
Pros and Cons: A Side-by-Side Comparison
Comparison Table: Small Molecule Drugs vs Gene Replacement
When it comes to treatment options, small molecule drugs and gene replacement therapies each have their strengths and limitations. The choice often depends on the patient's condition, access to care, and long-term management needs.
| Feature | Small Molecule Drugs | Gene Replacement Therapy |
|---|---|---|
| Administration | Oral pills or subcutaneous injection | IV infusion, subretinal injection, or intracranial delivery |
| Specificity | Lower; may affect multiple pathways | High; targets the specific genetic defect |
| Cost Considerations | Lower overall costs – with development typically 25–40% less expensive and a median ICER of $108,304 | Much higher costs – with a median ICER of $228,286 and up to tenfold higher costs |
| Bioavailability | Can cross the blood–brain barrier, aiding treatment for neurological conditions | Limited delivery to the CNS and skeletal muscle remains a challenge |
| Dosing Frequency | Daily dosing | Designed as a one-time or infrequent treatment |
| Manufacturing Scalability | High; benefits from fast, reproducible chemical synthesis processes | Challenging due to complex production logistics and limited vector supply |
| Immune Response Risk | Low risk of immunogenicity | High risk; immune responses (e.g., neutralizing antibodies to viral vectors) may occur |
Small molecule drugs primarily manage symptoms and slow disease progression but don't address the root cause of genetic conditions. These treatments require ongoing doses, which can be easier to manage financially but demand long-term adherence. On the other hand, gene replacement therapies aim to correct the genetic defect itself, potentially offering long-lasting or even permanent effects after just one treatment.
"Small molecules remain the majority [of approvals], which is relevant for supply‐chain risk and time‐to‐clinic considerations."
- SK pharmteco
Manufacturing processes also differ significantly. Small molecule drugs benefit from well-established chemical synthesis methods that are scalable and efficient. Gene therapies, however, involve intricate logistics, including cold-chain storage, specialized surgical delivery, and limited vector availability. These factors, combined with risks like immune responses and genotoxicity, make production and re-dosing more complicated.
Impact on Patient Access and Long-Term Care
The cost and delivery methods of these therapies play a major role in shaping patient access and ongoing care.
Small molecule drugs are generally less expensive to develop, with costs that are 25–40% lower than those of gene therapies. Their affordability extends to patients, with a median incremental cost-effectiveness ratio (ICER) of $108,304 compared to $228,286 for biologics. For example, Glybera, the first approved gene therapy in Europe, was withdrawn in 2017 due to its high cost and limited demand. Similarly, newer therapies like Casgevy and Lyfgenia face hurdles with reimbursement and the availability of specialized treatment facilities.
"Biologics are often ten times more expensive for patients than small molecule drugs. This can complicate treatment approval, insurance coverage and cause copay difficulties."
- Phil McHale, Consultant and former Director of Product Management, MDL
These cost disparities also influence long-term management. Small molecule drugs offer flexibility with adjustable dosing and are typically stable at room temperature. In contrast, gene therapies, though designed as one-time treatments, require rigorous long-term monitoring and often need refrigeration or cold-chain logistics for storage and delivery.
Ultimately, the choice between these options depends on the disease, the patient’s specific needs, and healthcare system capabilities. With over 95% of rare disease patients still lacking effective therapies, both approaches remain vital in addressing unmet medical needs.
Clinical Results and Patient Outcomes
Clinical Results: Small Molecule Drugs
Small molecule drugs have proven effective in managing rare genetic diseases, though their primary focus tends to be on slowing disease progression rather than curing the root cause. For example, in Type 1 Spinal Muscular Atrophy (SMA), the oral small molecule risdiplam delivered impressive results. Over a 36-month follow-up, it showed a 78% reduction in death rate compared to nusinersen, as well as an 81% reduction in the combined rate of death or permanent ventilation.
Patients receiving risdiplam also experienced substantial improvements in motor function. Specifically, they had a 45% higher rate of motor milestone responses on the Hammersmith Infant Neurological Examination (HINE-2) and a 186% higher rate of achieving at least a 4-point increase on the CHOP-INTEND score. Additionally, risdiplam was associated with a 57% lower incidence of serious adverse events compared to nusinersen.
Other rare conditions have also benefited from small molecule therapies. For acute hepatic porphyria (AHP), Givlaari reduced the annual porphyria attack rate from 12.5 to 3.2 in treated patients compared to the placebo group. Similarly, Oxlumo enabled 84% of patients with primary hyperoxaluria type 1 (PH1) to achieve normal or near-normal urinary oxalate levels within six months of treatment.
While these drugs are highly effective in managing symptoms and improving quality of life, gene replacement therapies aim to address the genetic root of these conditions, offering even more dramatic outcomes.
Clinical Results: Gene Replacement Therapy
Gene replacement therapies stand out for their ability to directly correct the underlying genetic defects, often resulting in transformative outcomes. For instance, Zolgensma, an AAV9-based therapy for SMA, achieved a 100% survival rate in its Phase 1 trial, compared to only 8% in historical cohorts.
In the realm of blood disorders, gene therapies have also delivered remarkable results. Zynteglo enabled approximately 89% (32 out of 36) of beta-thalassemia patients in Phase 3 trials to achieve transfusion independence. For sickle cell disease, Lyfgenia resolved severe vaso-occlusive crises in 94% of patients during the 6–18 months post-infusion, with 88.2% of patients experiencing no vaso-occlusive events at all.
However, gene therapies are not without risks. For example, Sarepta Therapeutics reported three patient deaths from acute liver failure following AAV gene therapy for muscular dystrophy in 2025. This led to a clinical hold on all of Sarepta's gene therapy trials and the suspension of shipments for their approved product, Elevidys. These incidents underscore the critical need for ongoing safety monitoring, even as gene therapies deliver high efficacy rates.
These findings highlight the importance of tailoring treatments to individual patient profiles. By carefully matching therapies to patients' specific needs, clinicians can maximize benefits while minimizing risks.
Matching Therapies to Individual Patients
What Determines the Best Treatment Choice
When it comes to treating genetic disorders, tailoring therapies to the specific needs of each patient is essential for achieving the best outcomes. Several factors influence which treatment option will work best.
First, the type of mutation plays a major role. For recessive monogenic disorders, where the problem stems from a missing or malfunctioning gene, gene replacement therapy can be highly effective. By introducing a functional version of the gene, this approach restores the missing activity. On the other hand, dominant genetic disorders or those caused by toxic gain-of-function mutations may respond better to small molecule drugs, which can target and manage the harmful effects directly.
The stage of the disease is another critical factor. For many degenerative conditions, early intervention is key. Gene therapy often works best when administered before symptoms appear, as it can halt or slow disease progression at its root. If symptoms have already begun, small molecule drugs might be more suitable for managing the condition as it advances.
Biological constraints also come into play. For example, patients with pre-existing immunity to viral vectors like AAV (used in gene replacement therapy) may not be eligible for this treatment. Small molecule drugs, however, don’t face this limitation. Similarly, the size of the gene involved can be a barrier - AAV vectors have a limited capacity, making them unsuitable for very large genes. Even within the same disease, differences in how it presents clinically may require a treatment plan tailored to the patient’s unique genetic makeup.
These complexities highlight the importance of precision medicine, where treatments are carefully matched to each individual’s genetic profile.
RareLabs' Personalized Drug Discovery Programs
RareLabs has developed a cutting-edge approach to tackle these challenges. By using patient-derived induced pluripotent stem cells (iPSCs), they create personalized cellular models. These models allow researchers to test various treatment options - such as small molecule drugs, antisense oligonucleotide therapy, and gene replacement therapy - on a patient’s own cells before deciding on the best course of action.
To ensure accuracy, RareLabs integrates CRISPR-corrected controls and karyotyping, setting precise baselines for their experiments. They also gather individualized natural history data to identify meaningful clinical outcome measures. This approach is especially valuable for ultra-rare conditions, which affect fewer than 1 in 1,000,000 individuals, where traditional clinical trials are often impractical.
What sets RareLabs apart is their commitment to transparency and patient engagement. Patients receive progress updates in plain language and have direct access to the science team throughout the discovery process, ensuring they remain informed and involved every step of the way.
Conclusion: Selecting the Right Treatment Approach
What Patients and Families Should Know
When it comes to rare diseases, families often face a mix of options, from gene therapies designed to address genetic defects to small molecule drugs that manage symptoms while waiting for gene therapy to become accessible.
With around 80% of rare diseases having a genetic origin, choosing the right treatment is highly specific to each case. Factors like detailed genetic testing and immune status (e.g., checking for neutralizing antibodies against viral vectors) play a crucial role. Gene replacement therapies, for instance, are effective only for certain mutations. A notable example is Luxturna, which targets biallelic RPE65 mutations. Additionally, families should consider the long-term commitment these therapies require. Many patients who undergo gene therapy join 15-year follow-up registries to monitor potential delayed effects, making it essential to balance upfront costs with ongoing care needs.
"Completely curing patients is obviously going to be a huge success, but it's not [yet] an achievable aim in a lot of situations." - Julie Crudele, Neurologist and Gene Therapy Researcher, University of Washington
Specialized centers are often the only places where advanced gene therapies are available, typically limited to certified academic research hospitals. Understanding these clinical and logistical factors is crucial for families making treatment decisions. It's also important to weigh financial considerations: therapies like Zolgensma come with a $2.1 million upfront cost, while small molecule treatments such as Spinraza cost about $750,000 in the first year and $375,000 annually thereafter.
How RareLabs Supports Treatment Development
RareLabs steps in to simplify this complex landscape by offering personalized drug discovery programs. These programs evaluate various therapeutic options - such as small molecule drugs, antisense oligonucleotide therapy, and gene replacement therapy - using patient-derived induced pluripotent stem cells. This approach is especially valuable for ultra-rare conditions affecting fewer than 1 in 1,000,000 individuals, where traditional clinical trials are not practical. By tailoring treatments to each patient’s unique genetic profile, RareLabs pushes forward the possibilities of personalized medicine in rare disease care.
FAQs
How do I know if I’m eligible for gene replacement therapy?
Eligibility for gene replacement therapy varies depending on your medical condition and whether the therapy has received approval. Many of these treatments are accessible only through clinical trials, which often have strict requirements. These can include factors like age, the severity of the disease, specific genetic markers, and your general health. For therapies approved by the FDA, eligibility usually hinges on having a confirmed diagnosis of the targeted genetic condition. To find out if you qualify, it’s best to consult with a healthcare provider who can guide you through the criteria for a particular therapy.
Can gene therapy be repeated if it wears off or doesn’t work?
Gene therapy can indeed be repeated if its effects diminish over time or if it doesn’t achieve the desired results initially. How long the therapy lasts can vary - some treatments may remain effective for more than a decade, but they aren’t always permanent. The possibility of repeating the therapy depends on several factors, including the type of vector used to deliver the genes, the body’s immune response, and the specific condition being treated. Researchers are continually working on improving gene editing techniques and delivery methods to make re-administration more practical when necessary.
Which option is safer long term: small molecules or gene therapy?
The long-term safety of small molecules and gene therapy remains under investigation. Small molecules tend to be viewed as safer because they have undergone extensive clinical testing and have well-documented safety profiles. That said, side effects are still possible. On the other hand, gene therapy, while offering exciting possibilities, is a newer approach. It carries potential risks, such as immune system reactions or unintended genetic alterations.
At present, the evidence leans toward small molecules being safer over the long haul. However, ongoing advancements in gene therapy are steadily improving its safety profile.



