Top 6 Therapeutic Approaches for Rare Genetic Conditions
Six modern treatments for rare genetic disorders: small molecules, ASOs, gene therapy, genome editing, combos, and AI-personalized drugs.

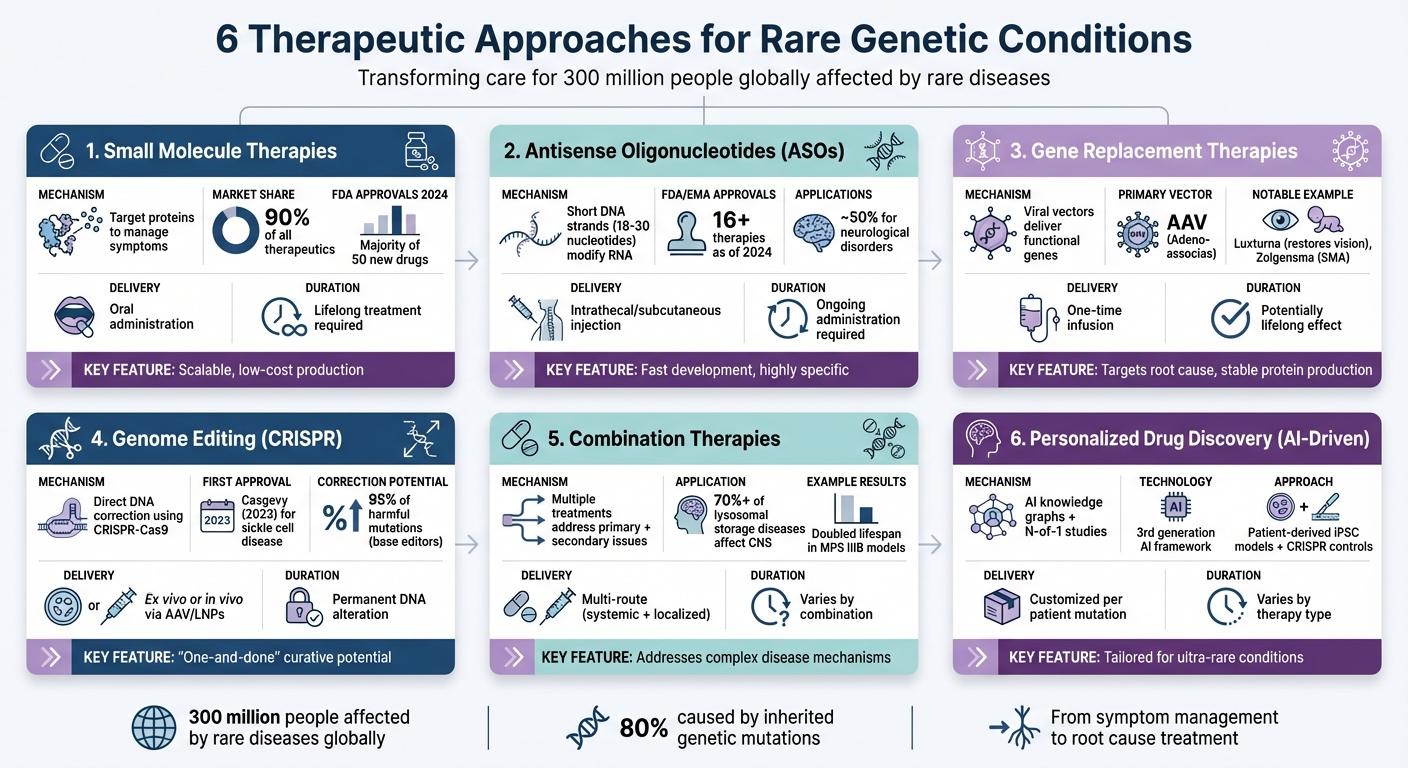
Rare genetic conditions affect 300 million people globally, with 80% caused by inherited mutations. While traditional treatments face economic and technical challenges, advancements in science are offering new possibilities. Here are six approaches transforming care for these conditions:
- Small Molecule Therapies: Target proteins to manage symptoms or slow disease progression, often using oral drugs.
- Antisense Oligonucleotides (ASOs): Short DNA strands that modify RNA to address specific genetic defects.
- Gene Replacement Therapies: Use viral vectors to deliver functional genes, offering lasting benefits for monogenic disorders.
- Genome Editing: Tools like CRISPR fix DNA errors directly, providing permanent solutions for some conditions.
- Combination Therapies: Multiple treatments address both primary genetic issues and related complications.
- Personalized Drug Discovery: AI-driven programs create patient-specific treatments for ultra-rare conditions.
These methods are redefining possibilities for treating rare diseases, aiming to address the root causes rather than just symptoms.
Comparison of 6 Therapeutic Approaches for Rare Genetic Conditions
1. Small Molecule Therapies
Mechanism of Action
Small molecules work by targeting proteins rather than genes. Their small size allows them to penetrate cells effectively, where they bind to proteins and modify their function. These compounds can act in several ways: as inhibitors that block harmful protein activity, as activators that enhance beneficial protein actions, or as stabilizers that help proteins maintain their proper structure.
A cutting-edge method called PROteolysis TArgeting Chimeras (PROTACs) takes this a step further. PROTACs utilize the ubiquitin-proteasome system to degrade problematic proteins entirely. This approach represents a major shift in treating diseases caused by malfunctioning proteins.
This ability to target proteins precisely forms the foundation of their use in addressing symptoms of rare genetic diseases.
Applications in Rare Genetic Conditions
Small molecules are often used to manage symptoms in rare genetic disorders, slowing disease progression and reducing the overall burden of the condition. They currently make up about 90% of all therapeutics available on the market. In 2024, the FDA approved 50 new drugs, with small molecules comprising the majority of these approvals.
Researchers are also leveraging AI-driven graph models to repurpose existing small molecules for ultra-rare conditions. This strategy speeds up development by building on existing toxicology and clinical data.
Advantages and Limitations
Small molecules come with several benefits, especially for rare diseases. They are often taken orally, which is convenient and reduces the need for frequent hospital visits. Their production is scalable, and their relatively low cost makes them ideal for clinical studies, even in very small patient populations.
"Small molecules remain the majority [of approvals], which is relevant for supply-chain risk and time-to-clinic considerations." - SK pharmteco
However, small molecules aren't without challenges. They can sometimes bind to unintended targets, causing side effects or toxicity. They are also prone to drug interactions and can only target about 3,000 of the 20,000 human proteins considered "druggable". Unlike gene therapies, which aim to provide a one-time cure, small molecules usually require ongoing treatment throughout the patient's life.
These targeted approaches highlight the growing precision in treating rare diseases.
A Breakthrough in Medicine: Personalized Gene Editing to Save KJ
2. Antisense Oligonucleotide (ASO) Treatments
ASO treatments represent a promising approach to addressing rare genetic disorders by targeting specific genes with precision.
Mechanism of Action
Antisense oligonucleotides are short, synthetic DNA strands, typically 18–30 nucleotides in length, designed to bind to specific RNA sequences within cells. This process relies on Watson-Crick base pairing, the same mechanism that governs DNA structure.
Once bound to their target RNA, ASOs can act in several ways: they may recruit RNase H to degrade the mRNA, alter splicing by skipping or including specific exons, or block translation by physically preventing ribosome access.
Chemical modifications, such as phosphorothioate backbones and 2'-O-methoxyethyl groups, significantly improve ASO stability, extending their half-life from mere minutes to several days. This added durability has turned ASOs into practical therapeutic tools.
Applications in Rare Genetic Conditions
ASOs have shown particular promise in treating neurological disorders, which account for nearly half of all rare conditions. As of 2024, at least 16 ASO-based therapies have been approved by the FDA or EMA for genetic disorders.
- Nusinersen (Spinraza): Approved in December 2016, this therapy combats Spinal Muscular Atrophy by promoting the inclusion of exon 7 in SMN2 mRNA, compensating for the missing SMN1 gene.
- Tofersen (Qalsody): Approved in 2023, it targets ALS caused by mutations in the SOD1 gene, degrading the toxic SOD1 mRNA.
- Eteplirsen (Exondys 51): This therapy addresses Duchenne Muscular Dystrophy by skipping exon 51 in the dystrophin gene, restoring the reading frame and enabling the production of a functional, albeit shorter, protein.
ASOs have even made personalized medicine possible. One groundbreaking example is Milasen, developed in 2018 for Mila Makovec, a child with Batten disease caused by a unique genetic mutation. Similarly, in August 2024, a patient with KIF1A-Associated Neurological Disorder received a custom ASO therapy from the n-Lorem Foundation. Over nine months of treatment, the patient experienced clinical improvements and stable cognitive function, according to research led by Wendy K. Chung at Columbia University Irving Medical Center.
Advantages and Limitations
ASOs offer several benefits but also come with challenges, particularly in delivery.
One of the biggest advantages is the speed of development. Since ASO design relies primarily on understanding mRNA sequences rather than complex protein structures, the development timeline is much faster than for protein-targeted drugs.
"Since ASO design relies principally upon knowledge of mRNA sequence, the bench to bedside pipeline for ASOs is expedient compared with protein-directed drugs." - Theodore P. Rasmussen, Department of Pharmaceutical Sciences, University of Connecticut
ASOs are highly specific, requiring only 13–15 consecutive nucleotides to target RNA effectively. They can either increase or decrease protein production as needed, and they avoid the permanent DNA alterations associated with gene editing, offering a safer alternative.
However, delivery remains a significant hurdle. ASOs are large and negatively charged, making it difficult for them to enter cells without help. Without chemical modifications, they are vulnerable to enzymatic degradation, and some modifications may cause side effects like liver toxicity or inflammation. Unlike one-time gene therapies, ASOs require ongoing administration throughout a patient’s life. Reaching tissues beyond the liver, such as the brain and muscles, is particularly challenging. New delivery technologies, including exosomes and lipid-albumin nanoparticles, are being explored to address these issues.
"These advancements not only enhance the precision and efficacy of antisense oligonucleotide therapies but also challenge researchers to innovate in overcoming delivery barriers, like hard-to-reach tissues and organs." - CAS Science Team
3. Gene Replacement Therapies
Gene replacement works by introducing a correct version of a gene into cells to restore proper protein function. To achieve this, scientists use modified viruses - primarily Adeno-associated virus (AAV) or Lentivirus - as carriers to deliver the therapeutic gene into the cell nucleus. These viral vectors are carefully engineered to remove harmful elements, ensuring they are safe for transporting genetic material. AAV vectors, in particular, are favored because they are safe, cause minimal immune reactions, and deliver stable gene expression without integrating into the patient’s chromosomes, which lowers the risk of unintended genetic changes.
Mechanism of Action
The process involves the viral vector delivering a functional gene that enables continuous protein production. Unlike antisense oligonucleotides (ASOs), gene replacement therapy can provide lasting benefits in non-dividing cells with just a single dose. This makes it especially effective for recessive monogenic disorders - conditions caused by a single faulty gene - where even a modest restoration of protein levels can lead to meaningful clinical improvements.
This precise approach has laid the foundation for successful treatments targeting rare conditions.
Applications in Rare Genetic Conditions
Gene replacement therapy has shown extraordinary results in treating conditions like eye diseases, neuromuscular disorders, and blood disorders. One standout example is Luxturna (Voretigene neparvovec-rzyl), an AAV-based therapy approved for Leber Congenital Amaurosis. This treatment restores vision in patients with inherited retinal disease. In a phase 3 trial, all 20 treated patients retained improved vision three years post-treatment. Patients reported life-changing visual improvements, though the cost of treatment is steep - about $425,000 per injection, or nearly $1 million for both eyes.
For Hemophilia B, gene therapy has revolutionized care. In a Phase 3 trial with 54 men, participants received an infusion of 2 × 10¹³ genome copies per kilogram of an AAV5 vector carrying the Padua factor IX variant. This reduced the annual bleeding rate from 4.19 before treatment to 1.51 between months 7 and 18 after therapy. Similarly, Hemophilia A patients treated with Roctavian experienced a mean factor VIII activity increase of 41.9 IU per deciliter after one year, with a 98.6% reduction in the use of factor concentrates.
Another success story is Zolgensma, designed for Spinal Muscular Atrophy (SMA). Delivered via an AAV9 vector, this therapy provides a functional copy of the SMN1 gene. Before Zolgensma, infants with Type I SMA faced a 90% mortality rate by their first birthday. This therapy has been able to halt disease progression and restore motor function in patients who would otherwise face severe, life-threatening muscle weakness.
Advantages and Limitations
Gene replacement therapies come with transformative potential but also notable challenges.
One key advantage is the consistent protein production they enable. Unlike enzyme replacement therapies, which require frequent infusions and lead to fluctuating protein levels, gene therapies provide stable, ongoing protein expression. They also eliminate the risk of graft-versus-host disease, which can occur with donor bone marrow transplants.
"Gene therapy offers the potential for a cure by targeting the underlying cause rather than just managing symptoms." - Muhammad Umair, Medical Genomic Research Department, KAIMRC
However, there are hurdles to overcome. High doses of systemic AAV can lead to liver toxicity or microangiopathy, and immune system responses to the viral vectors may reduce their effectiveness. Delivering genes to specific tissues, like the central nervous system, remains a challenge, and controlling transgene expression is still limited. Additionally, this therapy is less effective for dominant genetic disorders, complex polygenic conditions, or diseases requiring genes too large for the viral vector to carry. The high cost of these treatments also makes them among the most expensive in the U.S..
"Completely curing patients is obviously going to be a huge success, but it's not [yet] an achievable aim in a lot of situations." - Julie Crudele, Neurologist and Gene Therapy Researcher, University of Washington
Despite these obstacles, the impact on patients’ lives has been extraordinary. Jean Bennett, an ophthalmologist at the University of Pennsylvania, observed:
"These people can now do things they never could have dreamed of doing, and they're more independent and enjoying life".
4. Genome Editing Therapies
Genome editing marks a shift from simply adding new genes to precisely correcting DNA errors. Unlike gene replacement therapies that introduce a fresh copy of a gene, genome editing directly fixes the faulty DNA sequence. The most recognized tool in this field is CRISPR-Cas9. This system uses a single-guide RNA (sgRNA) to direct the Cas9 enzyme to a specific DNA target, where it creates a double-strand break. The cell then repairs this break through one of two pathways: Non-Homologous End Joining (NHEJ), which often leads to small errors like insertions or deletions, or Homology-Directed Repair (HDR), which uses a template to make precise corrections.
This technology builds on earlier genetic approaches, offering new possibilities for more precise and lasting treatments.
Mechanism of Action
Advancements in genome editing now include tools like base and prime editors, which allow scientists to alter DNA without breaking both strands. Base editors can change a single DNA letter, such as converting cytosine (C) to thymine (T) or adenine (A) to guanine (G). This method could potentially fix around 95% of harmful mutations cataloged in ClinVar. On the other hand, prime editors can introduce more complex changes, like point mutations, insertions, or deletions, though they tend to work less efficiently.
Delivery methods for these tools depend on the target tissue and the size of the editor. Recombinant adeno-associated viruses (rAAV) and lipid nanoparticles (LNPs) are commonly used. While rAAV has a packaging limit of less than 4.7 kilobases, LNPs can handle larger payloads, offering more flexibility.
Applications in Rare Genetic Conditions
In late 2023, the FDA approved Casgevy (exagamglogene autotemcel), developed by Vertex Pharmaceuticals and CRISPR Therapeutics. This was the first CRISPR-based therapy to hit the market, targeting sickle cell disease and beta-thalassemia. It works by editing the BCL11A gene in hematopoietic stem cells to reactivate fetal hemoglobin. This ex vivo approach involves removing the patient’s stem cells, editing them in a lab, and then reinfusing them.
For in vivo editing, where the therapy is delivered directly into the body, Editas Medicine's EDIT-101 is a notable example. This therapy targets the IVS26 mutation in the CEP290 gene, which causes Leber Congenital Amaurosis type 10 (LCA10). Delivered via an AAV5 subretinal injection, the Phase 1/2 BRILLIANCE trial showed improved photoreceptor function in 11 out of 14 participants. Meanwhile, Verve Therapeutics is exploring base editing to treat familial hypercholesterolemia by targeting the PCSK9 gene in the liver.
In May 2025, researchers from the Children's Hospital of Philadelphia and the University of Pennsylvania achieved a groundbreaking milestone. They published results in the New England Journal of Medicine detailing the first personalized gene-editing therapy for an infant. The patient, KJ Muldoon, was diagnosed with carbamoyl-phosphate synthetase 1 (CPS1) deficiency, a rare urea cycle disorder affecting one in a million babies. Without treatment, toxic ammonia buildup would cause severe brain damage or death. The team, led by Dr. Rebecca Ahrens-Nicklas and Dr. Kiran Musunuru, developed a tailored base-editing therapy delivered via LNPs. By nine months old, KJ was reaching developmental milestones like sitting up and rolling over.
"Ultimately we hope this has set a precedent where we have firmly entered a world of genetic cures - CRISPR cures - on demand. I think we can say: This is the year when CRISPR-on-demand is truly born." - Fyodor Urnov, Scientific Director of the Innovative Genomics Institute at UC Berkeley
These cases highlight how genome editing is paving the way for personalized treatments, especially for rare and ultra-rare conditions.
Advantages and Limitations
Genome editing offers the promise of a one-time cure. Unlike therapies like antisense oligonucleotides, which require ongoing treatment, genome editing permanently alters the DNA. Techniques like base and prime editing also reduce risks like chromosomal translocations and stress responses linked to double-strand breaks. Additionally, rAAV-based gene expression has been shown to last over a decade in some clinical scenarios.
"Gene editing therapy emerges as a promising approach to craft curative and lasting treatments for these patients, often referred to as 'one-and-done' therapeutics." - Júlia-Jié Cabré-Romans and Raquel Cuella-Martin, McGill University
However, challenges remain. Off-target effects, whether Cas-dependent or independent, pose safety concerns. Delivery is another hurdle, especially for larger tools like base and prime editors that exceed AAV’s size limits. Researchers are exploring solutions like "split-intein" systems, which assemble proteins from two separate vectors, or "RNA trans-splicing" methods. Immune reactions to bacterial Cas proteins and the limited application of HDR-based editing to dividing cells also restrict broader use.
Despite these obstacles, the field continues to move forward. Reflecting on the groundbreaking treatment of KJ Muldoon, Dr. Rebecca Ahrens-Nicklas remarked:
"The first time you're putting a new drug into a baby is scary. No one has done this before. No one has developed a personalized gene-editing therapy for an infant."
As researchers refine these techniques, experts believe the barriers to personalized gene editing will diminish significantly in the coming years.
5. Combination Therapeutic Approaches
Treating rare diseases often requires more than a single approach. While single-modality treatments can address the primary defect, many conditions demand a combination of therapies to tackle both the root genetic issue and secondary complications like neuroinflammation or oxidative stress. For example, pairing enzyme replacement therapy (ERT) with small molecules or combining gene therapy with bone marrow transplantation offers a way to address these challenges from multiple angles.
Mechanism of Action
Combination therapies work through multiple mechanisms to combat disease. Take lysosomal storage diseases (LSDs) as an example - these conditions affect the central nervous system in over 70% of cases. Researchers often combine ERT, which breaks down accumulated substrates, with substrate reduction therapy (SRT), which reduces the production of new substrates. To ensure these treatments reach both peripheral organs and the brain, multiple delivery routes are used. For instance, systemic delivery of a viral vector can help reduce immune responses, making localized treatments more effective.
These carefully designed methods form the backbone of the clinical advancements discussed below.
Applications in Rare Genetic Conditions
Recent trials highlight how combination therapies are making a difference in rare diseases. Between 2024 and 2025, a phase 1/2 clinical trial (NCT04669535 and NCT06614569) tested a dual-vector approach on nine children with Tay-Sachs and Sandhoff diseases (GM2 gangliosidosis). This treatment involved a single administration of two vectors - rAAVrh8-HEXA and rAAVrh8-HEXB - delivered through bi-thalamic injections, intra-cisterna magna, and intrathecal infusions. Results were promising: cerebrospinal fluid HexA enzyme activity reached about 13% of normal levels (0.59 nmol·h⁻¹·ml⁻¹), and total serum Hex activity hit 40 nmol·h⁻¹·ml⁻¹, doubling the lower limit of the normal range. Remarkably, infantile patients were able to maintain oral feeding without aspiration until 3–3.5 years of age, a significant improvement over historical outcomes.
In another study, researchers explored treating Sanfilippo's Syndrome (MPS IIIB) in mouse models using both intracranial and systemic delivery of AAV and lentiviral vectors expressing the NAGLU enzyme. This approach nearly doubled the mice's lifespan and corrected motor dysfunction, hearing loss, and disrupted circadian rhythms. Similarly, in mouse models of Infantile Neuronal Ceroid Lipofuscinosis (INCL), combining gene therapy with a glial-specific anti-inflammatory small molecule improved motor function, extended lifespan, and completely eliminated seizures.
Advantages and Limitations
Combination therapies offer benefits that single treatments often can't achieve. For instance, in Krabbe disease models, combining gene therapy with bone marrow transplantation nearly eliminated neuroinflammation, while bone marrow transplantation alone provided limited results. Additionally, small molecule drugs capable of crossing the blood-brain barrier can address neurological symptoms that standard ERT cannot.
"By optimizing routes of delivery, therapeutic timing, and targeting secondary disease mechanisms, combination therapy represents the future for LSD treatment." - Shannon L. Macauley, PhD, Washington University
However, these approaches come with challenges. Not all combinations work universally. For example, pairing gene therapy with bone marrow transplantation in certain MPS IIIB models unexpectedly worsened motor performance and reduced lifespan. Procedures like stem cell or bone marrow transplantation often require intensive conditioning regimens, which can be risky for vulnerable patients. In the GM2 gangliosidosis trial, some juvenile patients developed new or worsening dystonia within one to two months of treatment, leading to their exclusion from further participation. Additionally, participants experienced a more than tenfold increase in serum neutralizing antibody levels to the AAVrh8 capsid, requiring close immune monitoring and steroid doses ranging from 2 to 10 mg·kg⁻¹·day⁻¹.
This multi-faceted approach demonstrates the importance of tailoring treatments to individual needs, pushing the boundaries of personalized medicine for rare genetic conditions.
6. RareLabs Personalized Drug Discovery Program
Mechanism of Action
RareLabs is pushing the boundaries of personalized medicine with its cutting-edge drug discovery program. This program leverages a third-generation AI framework that combines biomedical research, scientific literature, and patient data into a unified knowledge graph. The goal? To uncover new connections between diseases and potential treatments.
The process begins with genome sequencing to pinpoint each patient's unique genetic defect. From there, scientists use an "N-of-1" study model to design, test, and produce customized therapies - tailored specifically for patients who have no other treatment options. This approach aligns with the FDA's recent shift in regulations, which now allows approvals based on a "plausible mechanism" of action instead of requiring the lengthy traditional clinical trial process.
Applications in Rare Genetic Conditions
RareLabs has developed a structured, three-step strategy to address rare genetic disorders:
- Patient-derived models: Using induced pluripotent stem cells (iPSCs) from the patient, researchers create models of the condition. These models are paired with CRISPR-corrected controls and undergo detailed karyotyping.
- Therapeutic screening: A range of treatment options - including small molecules, antisense oligonucleotide (ASO) therapies, gene replacement, and combination approaches - are screened to find the most effective solution for the specific mutation.
- Direct communication: Patients receive regular updates and can interact directly with the scientific team to stay informed about progress.
For individuals with splice-site mutations or other rare genetic variants, the program can develop splice-modulating ASOs tailored to the mutations identified during genome sequencing.
Advantages and Limitations
One of the standout benefits of this program is its focus on "derisked" drug candidates - those that already have a track record of safety or effectiveness in related conditions. This strategy not only reduces development risks but also speeds up the process. The AI-driven knowledge graph further enhances efficiency by identifying opportunities for drug repurposing and innovative therapeutic combinations.
That said, personalized drug discovery isn't without its hurdles. Costs and timelines are highly variable, making it challenging to predict expenses. Additionally, the complexity of creating patient-specific treatments can lead to lengthy development processes, and not all genetic mutations respond well to available therapies. While the FDA's updated regulatory pathway simplifies approvals for individualized treatments, navigating this process still demands meticulous documentation and ongoing safety checks.
Comparison Table
Genome editing and RNA-based therapies differ in how they work, how they are delivered, and the immune responses they trigger. Here's a breakdown of the key differences between these two therapeutic approaches for rare genetic conditions.
Genome editing works by directly altering DNA to make permanent changes at specific locations in the genome. CRISPR systems, for example, use a 20-base pair guide RNA to direct the Cas9 protein to the target DNA sequence. While this precision is powerful, any mistakes - like large deletions or chromosomal rearrangements - are irreversible. On the other hand, RNA-based therapies, such as antisense oligonucleotides (ASOs), target messenger RNA to adjust protein production without modifying the DNA itself. These effects are temporary, meaning repeated treatments are necessary.
Delivery methods also set these two apart. Genome editing tools often rely on viral vectors, such as adeno-associated viruses (AAV), which are limited to carrying genetic material under 4.7 kilobases. RNA-based therapies bypass this size limitation but face their own hurdles, like instability and low bioavailability of RNA molecules. To address these issues, chemical modifications or specialized delivery systems are often required.
Immune responses differ significantly between the two approaches. Genome editing can provoke immune reactions against bacterial proteins like Cas9 or the viral vectors used for delivery. Pre-existing antibodies to common viral vectors can also neutralize the therapy before it reaches its target. Meanwhile, RNA-based therapies may activate the innate immune system due to the synthetic nature of the oligonucleotides or cause infusion reactions when delivered via lipid nanoparticles. Newer delivery methods, such as GalNAc conjugation for liver-targeted therapies, have shown reduced toxicity compared to traditional lipid nanoparticle systems.
The table below highlights these differences:
| Factor | Genome Editing (CRISPR, Base Editors) | RNA-Based Therapies (ASOs, siRNA) |
|---|---|---|
| Primary Target | Genomic DNA | Messenger RNA or pre-mRNA |
| Duration of Effect | Permanent/Heritable | Temporary |
| Delivery Vehicle | Viral vectors (AAV, Lentivirus) or LNPs | Non-viral systems, LNPs, or direct oligonucleotides |
| Cargo Constraints | Limited (AAV max. 4.7 kb) | Minimal; limited by molecular stability |
| Immune Risk | Responses to viral capsids and bacterial proteins | Innate immune activation by oligonucleotides |
| Dosing Frequency | Potentially one-time treatment | Chronic, repeated administration |
| Manufacturing Scalability | Low; viral production is complex and costly | High; chemical synthesis is more scalable |
These distinctions underline the tailored approaches required for managing rare genetic diseases. Each method comes with its own set of challenges and advantages, shaping how they are used in different therapeutic contexts.
Conclusion
The six approaches outlined earlier mark a major shift in how rare genetic diseases are treated. Instead of just managing symptoms, these therapies aim to address the root causes of the diseases, offering the possibility of long-term improvement or even a cure. Recent advancements have shown real promise, with some patients experiencing life-changing outcomes like restored vision and children gaining the ability to walk independently.
Globally, around 300 million people are affected by rare diseases, with 80% stemming from inherited genetic abnormalities. This underscores the urgent need for effective treatments. As Siddhi Desai and colleagues from Ashokrao Mane College of Pharmacy aptly stated:
"With coordinated global efforts from research institutions, industry, and regulatory bodies, gene therapy has the potential to overcome existing challenges and achieve a transformative impact on modern medical practice".
Collaboration is already making a difference. For instance, in January 2026, the n-Lorem Foundation partnered with the EspeRare Foundation to expand access to individualized antisense oligonucleotide therapies in Europe. Their initial focus is Switzerland, where they aim to treat up to three nano-rare patients within the first year. Sarah Glass, Ph.D., Chief Operating Officer at n-Lorem Foundation, highlighted a key challenge:
"Individualized ASO therapies can only reach patients if the underlying regulatory and clinical frameworks are capable of supporting them".
Looking ahead, efforts must focus on reducing costs, enhancing quality control, and refining regulatory systems to ensure these therapies can reach those with ultra-rare conditions. High costs remain a significant obstacle, often halting progress on promising treatments. Overcoming these barriers will require continued collaboration among researchers, clinicians, patients, and regulatory bodies.
These advancements not only represent scientific progress but also bring renewed hope to millions of patients. The future of personalized treatments depends on sustained efforts and partnerships to make these life-changing therapies accessible to all who need them.
FAQs
How do doctors decide which therapy fits a specific mutation?
Doctors determine the best therapy by examining the genetic mechanism driving a disease. Through genetic testing, they pinpoint specific mutations, which helps guide treatment choices such as small molecule drugs, antisense oligonucleotides (ASOs), or gene replacement therapies. These treatments are designed to address the mutation's impact on gene function, ensuring they match the patient’s unique genetic profile. Advances in personalized medicine are making these targeted approaches increasingly effective for rare genetic conditions.
What are the biggest safety risks with gene therapy or CRISPR?
Gene therapy and CRISPR come with several safety challenges, including off-target effects, immune responses, and ethical dilemmas.
Off-target effects happen when unintended parts of the genome are modified, which can lead to harmful mutations. This is a significant concern because even small, unintended changes in DNA could disrupt important genes or pathways.
Immune responses are another risk. The body might react to the tools used for gene editing, such as delivery vectors or enzymes, triggering inflammation or other immune-related issues. These reactions could complicate treatment or reduce its effectiveness.
Then there are ethical concerns, especially with germline editing, which involves changes that can be passed down to future generations. Questions about consent and the long-term consequences of altering the human genome make this a particularly sensitive issue.
Researchers are actively working to address these challenges, focusing on improving precision, minimizing risks, and developing clear regulations to guide the responsible use of these technologies.
Why do some treatments need repeat dosing while others are one-time?
Some treatments need to be administered repeatedly because they don't offer a lasting solution to the underlying genetic issue or damaged cells. For instance, small molecule drugs and antisense oligonucleotide (ASO) therapies require continuous dosing to stay effective, as the body gradually eliminates them. On the other hand, gene therapies are designed as one-time treatments to permanently address the defective gene. However, even these may occasionally require follow-up if their effects wane over time.



