Rare Disease Treatment Checklist: 7 Things to Consider
Seven-step checklist to choose genetic therapies, use patient-derived models, confirm safety, and plan translation for rare diseases.

Treating rare diseases is complex, especially when they affect fewer people and lack approved therapies. Here's what you need to know to navigate treatment options effectively:
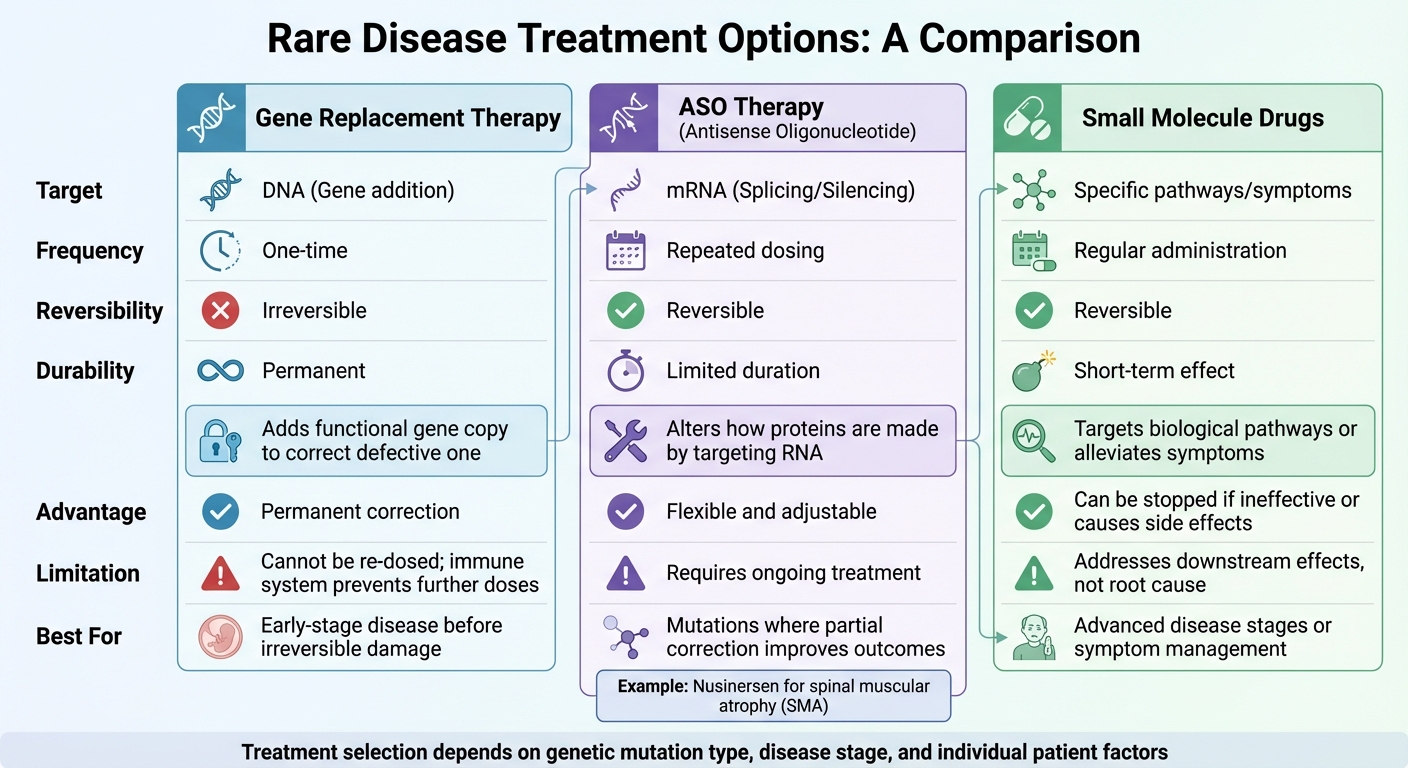
- Evaluate Treatment Options: Different therapies like gene replacement, small molecule drugs, and antisense oligonucleotide (ASO) therapy target specific needs. Each has unique benefits and limitations based on disease stage and mutation type.
- Use Patient-Derived Cell Models: Induced pluripotent stem cells (iPSCs) from patients provide accurate platforms for testing drugs tailored to genetic profiles.
- Complete Genetic Profiling: Tools like Whole Exome Sequencing (WES) and Whole Genome Sequencing (WGS) identify mutations, guiding precise treatment.
- Test Multiple Approaches: Combining therapies and testing in organoid models improves the likelihood of finding effective solutions.
- Validate Safety and Efficacy: Rigorous testing ensures treatments are safe and meet regulatory standards.
- Track Progress Clearly: Milestone reporting keeps families informed and engaged throughout the process.
- Plan for Clinical Translation: Early regulatory planning and long-term monitoring ensure successful treatment delivery.
These steps help families and researchers make informed decisions while addressing the challenges of developing treatments for rare genetic conditions.
Comparison of Gene Replacement Therapy, ASO Therapy, and Small Molecule Drugs for Rare Diseases
Tuesday's Daily Dose: FDA fast-tracks rare disease treatments and cancer genetic tests have limits
1. Assess Treatment Options for Your Specific Condition
Rare diseases are often caused by single-gene defects, making treatment highly specific to the genetic mutation and the stage of the disease. Options like gene replacement therapy, small molecule drugs, or antisense oligonucleotide (ASO) therapy are tailored to these factors.
These decisions are critical because some treatments, like gene therapy, are irreversible. Once administered, they may limit future gene-based options if they don't work as intended. Timing is equally crucial - patients must be in a "Goldilocks" zone, where they are affected enough to benefit but not so advanced that irreversible damage has already occurred. Below is a breakdown of how these treatments address different challenges.
Gene Replacement Therapy
Gene replacement therapy works by adding a functional gene copy to correct a defective one. As the NIH MedlinePlus Magazine explains, gene therapies aim to "fix" genetic mutations by replacing or altering faulty genes. This approach is permanent, as the new gene integrates into the DNA, eliminating the possibility of re-dosing.
This permanence is both an advantage and a drawback. Once the therapy is administered - usually through viral vectors - the immune system often prevents further doses. For conditions like Duchenne muscular dystrophy (DMD), the high doses required can increase the risk of side effects. Additionally, this therapy cannot reverse damage already present, so timing is essential to maximize its benefits.
Small Molecule Drugs
Small molecule drugs take a different route by targeting biological pathways or alleviating symptoms, rather than fixing the genetic defect itself. These treatments are reversible - if they don't work or cause side effects, you can stop taking them.
For patients with advanced disease stages, where genetic correction can't repair existing damage, small molecules can offer meaningful symptom management. They are also beneficial for mutations that aren't suitable for gene replacement or ASO therapies. However, the main trade-off is that small molecules usually address downstream effects rather than the root cause of the disease.
Antisense Oligonucleotide (ASO) Therapy
ASO therapy targets RNA, offering a middle ground between gene replacement and small molecule treatments. According to the American Society of Gene & Cell Therapy:
"ASO therapies act inside a cell to alter how proteins are made. ASOs can silence a gene so a protein is not made. It can also alter how mRNA is made to then change how a protein is made".
Unlike gene replacement, ASOs are not permanent and require repeated dosing, which allows for flexibility and adjustments if needed. For example, nusinersen, an ASO therapy for spinal muscular atrophy (SMA), modifies the SMN2 gene to boost production of the survival motor neuron protein. While repeated dosing is necessary, this approach offers adaptability that one-time gene therapies lack. ASOs are particularly effective for mutations where even partial correction can improve outcomes.
| Feature | Gene Replacement Therapy | ASO Therapy | Small Molecule Drugs |
|---|---|---|---|
| Target | DNA (Gene addition) | mRNA (Splicing/Silencing) | Specific pathways/symptoms |
| Frequency | One-time | Repeated dosing | Regular administration |
| Reversibility | Irreversible | Reversible | Reversible |
| Durability | Permanent | Limited duration | Short-term effect |
RareLabs provides a Personalized Treatment Assessment Plan to help determine which therapeutic approach - or combination - best suits your genetic profile and disease stage. This tailored evaluation helps guide your treatment decisions effectively.
2. Use Patient-Derived Cell Models for Drug Testing
Patient-derived induced pluripotent stem cells (iPSCs) offer a unique advantage: they mirror your complete genetic makeup, including any disease-causing mutations or modifiers. This makes them much more reliable than animal models, which often fail to predict outcomes in human clinical trials due to differences between species. With this level of accuracy, researchers can pinpoint mutation-specific effects more effectively.
To maximize the potential of these cells, researchers often pair them with CRISPR-corrected controls. By using CRISPR to revert a specific mutation to its healthy form - while keeping the rest of the genetic background unchanged - it becomes possible to isolate the mutation's impact. For example, in February 2026, researchers studying NGLY1 deficiency used this technique. They found that CRISPR-corrected controls restored normal neurodevelopment and mitochondrial function, confirming the mutation's direct role in the disease.
Creating iPSCs
The process starts with a simple skin or blood sample, which is reprogrammed into iPSCs using factors like Oct4, Sox2, Klf4, and Myc. This involves two key steps: first, erasing the cell’s original identity through DNA demethylation and chromatin remodeling; second, activating genes responsible for pluripotency to produce cells that can differentiate into various tissues.
However, the process isn’t without challenges. Over 70% of iPSC lines develop mitochondrial DNA mutations during reprogramming. Additionally, blood-derived iPSCs typically show fewer UV-related mutations compared to those from skin samples, which can influence the model’s accuracy. Adjusting reprogramming methods based on the tissue source improves reliability. Growth protocols must also account for genetic differences that affect how efficiently cells reprogram and differentiate.
Quality Control with CRISPR and Karyotyping
Quality control is critical when working with iPSCs. Karyotyping is performed multiple times - both after reprogramming and following any CRISPR edits - to detect chromosomal abnormalities that could interfere with drug testing. Researchers also use SNP arrays to identify copy number variations and long-range PCR to ensure CRISPR edits are precise and free of off-target effects.
RareLabs specializes in creating these patient-derived iPSC models, complete with CRISPR-corrected controls and thorough quality checks, including karyotyping. This ensures that the cells accurately reflect the disease state, making them an ideal platform for testing potential treatments. By using isogenic pairs as experimental controls, researchers can identify drugs that specifically target the mutation in question. These stringent measures help advance the development of personalized therapies.
3. Complete Genetic Profiling to Identify Mutations
Before starting treatment, it's crucial to identify the mutation causing the condition. Two key tools for this are Whole Exome Sequencing (WES) and Whole Genome Sequencing (WGS). WES focuses on 1–2% of the genome but captures around 85% of known disease-causing mutations. On the other hand, WGS examines the entire DNA sequence, including non-coding regions, structural variants, and repeat expansions - areas that WES often overlooks.
Choosing between WES and WGS depends on the specific case. WES is typically the first step due to its lower cost and strong ability to detect pathogenic variants. If WES doesn't provide clear results, WGS is the next step, especially for conditions involving repeat expansions (like Huntington's disease) or regulatory mutations in non-coding regions. The American College of Medical Genetics and Genomics recommends both WES and WGS as primary or secondary testing options for developmental delays and intellectual disabilities. Pinpointing the exact mutation is essential for accurate diagnosis and effective treatment planning.
Interpreting the data accurately is just as important. Patients often endure a long diagnostic journey - averaging seven years - before receiving answers. WGS generates about 3 million mutations per individual, compared to WES's 100,000. Most of these mutations are harmless, so tools like population databases (e.g., gnomAD) help filter out benign variants to focus on the pathogenic ones.
Performing trio analysis (sequencing the patient and both parents) can significantly enhance diagnostic accuracy by identifying de novo mutations - those not inherited from either parent. For example, a study at the UCLA Care and Research in Neurogenetics Clinic reviewed 316 patients between 2019 and 2024. Of these, 78% underwent genetic testing, leading to the identification of pathogenic or likely pathogenic variants in 152 cases. In 36 patients diagnosed directly through the clinic, the findings prompted changes in medical management.
Revisiting genetic data every 1–2 years is crucial to account for new discoveries. Among positive WES results, 23% involve genes identified in the last two years, with 7% tied to entirely new gene discoveries. As genetic databases expand and hundreds of new gene-disease associations are uncovered annually, variants initially labeled as "uncertain significance" are often reclassified as either pathogenic or benign. This evolving knowledge plays a key role in refining diagnoses and guiding personalized treatment strategies.
4. Test Multiple Treatment Approaches
Once a mutation is identified, the next step is to explore various treatment strategies to find the most effective solution. Using patient-derived models, researchers can test different options tailored to the specific mutation. For instance, structural deletions in the DMD gene may respond to exon-skipping antisense oligonucleotides (ASOs), while deep intronic variants that create novel splice sites often require custom-designed ASOs.
Each type of mutation benefits from a distinct treatment approach:
- Gene replacement therapy: This method introduces a functional gene as a one-time correction targeting the root cause.
- ASOs: These bind to mRNA, offering flexibility in dosing and adaptability to specific needs.
- Small molecules: Identified through high-throughput screening, these can reverse disease phenotypes.
Patient-derived organoids have revolutionized drug testing, cutting development time from years to just a couple of months. For example, in a study on Duchenne muscular dystrophy, researchers worked with cardiac organoids from a patient missing exons 46–53. They tested an ASO designed to skip exon 45, successfully restoring dystrophin levels to those seen in healthy organoids and reestablishing normal calcium rhythms. Similarly, for two siblings with a deep intronic mutation, personalized ASOs corrected DMD splicing and restored protein expression in their cardiac organoids.
Combining Multiple Treatment Strategies
Combining different treatment methods can enhance results even further. Patient-derived organoids provide a platform for testing multiple approaches simultaneously. For example, researchers can screen ASOs targeting various transcript regions - such as start sites or splice junctions - while also testing small molecule libraries within the same system. This integrated strategy is especially critical for ultra-rare diseases where traditional clinical trials are not practical.
"The ability to customize ASO designs to individual patients has major implications for personalized therapeutics." – Nature
Gene therapy, while offering a permanent solution, comes with risks. Once administered, it cannot be undone, and unsuccessful attempts may exclude patients from future trials. Testing both gene therapy and ASO options in organoid models ensures the most promising treatment path is identified without limiting future possibilities.
5. Validate Safety and Efficacy Before Clinical Use
Before any therapy can reach patients, it must undergo rigorous testing to confirm it is both safe and effective. The FDA requires that product quality controls are established early in development, especially for therapies on accelerated timelines.
This validation process includes in vitro (lab-based), in vivo (animal-based), and in silico (computer-simulated) studies to create a thorough safety profile. Take onasemnogene abeparvovec, for instance - a treatment for spinal muscular atrophy approved in May 2019. Its approval relied on external control data from the Pediatric Neuromuscular Clinical Research database to evaluate efficacy in a single-arm trial. Similarly, bluebird bio used a retrospective chart review of patients with cerebral adrenoleukodystrophy (from 1988 to 2010) as a natural history control to support their gene therapy approval.
"Sponsors should consider all scientifically relevant and regulatorily acceptable study/testing options available (e.g., microphysiologic systems, relevant animal models, etc.) to diminish both the time and number of participants required at all stages of the clinical program." – Premier Research
For gene therapies, the FDA mandates 15 years of follow-up to monitor for delayed adverse effects like malignancies or persistent infections. Between 2016 and 2023, 12 gene therapy products targeting rare diseases were approved, with five programs utilizing natural history data as their primary control group. This approach is particularly helpful when traditional placebo-controlled trials would be unethical or when patient populations are too small for randomized studies.
Standardized Validation Protocols
Beyond comprehensive validation, standardized protocols are critical to ensure reliable data and regulatory compliance. Trials must adhere to Good Clinical Practice (GCP) standards, such as ICH E6(R2), which focus on protecting participants and generating credible results. Clinical outcome assessments also need to demonstrate variability, sensitivity, reliability, and relevance to patients.
The Plausible Mechanism Framework provides a structured method for validating the safety and efficacy of individualized therapies, especially those targeting genetic conditions. For example, in the development of beremagene geperpavec (a treatment for dystrophic epidermolysis bullosa), researchers used a randomized, double-blind, intrasubject placebo-controlled design. This involved treating one wound on a participant with the therapy while using a placebo on a comparable wound, ensuring high-quality data while reducing the burden on patients.
Early engagement with regulatory bodies is another cornerstone of successful validation. Pre-IND meetings allow sponsors to discuss development plans, study designs, and confirmatory evidence requirements before starting pivotal trials. As highlighted in regulatory guidance:
"Regulatory engagement early and often." – Advances in Therapy
This proactive strategy ensures that endpoint selection and clinical measurement plans align with FDA expectations, minimizing the risk of delays or costly trial redesigns later in the process.
6. Track Progress with Clear Milestone Reporting
Developing treatments for rare diseases often takes years, making clear milestone reporting crucial to keep everyone on the same page. Without this transparency, families can feel lost in the maze of technical data, which may lead to confusion and disengagement.
In January 2026, Agios Pharmaceuticals set an example by sharing a detailed roadmap under CEO Brian Goff. This report highlighted key milestones such as the late January 2026 U.S. commercial launch of AQVESME™ for thalassemia, a planned pre-sNDA meeting for sickle cell disease in Q1 2026, and the anticipated start of a Phase 1b trial for PKU in the first half of the year. By tying progress to specific timelines, they provided clarity and focus.
When tracking milestones, it’s important to monitor key points like natural history baselines, regulatory designations, first-in-human trials, and required long-term follow-ups. For example, the FDA mandates a minimum 5-year follow-up for adeno-associated viral vectors and at least 15 years for integrating vectors.
Plain-Language Updates
To help stakeholders understand complex milestones, translating technical terms into everyday language is essential. Plain-language summaries make dense scientific data easier for families to grasp. Take Luxturna’s Phase III trial for retinal dystrophy as an example: researchers used a "multi-luminance mobility test" to measure efficacy. To make these results relatable, they showed how this advanced test correlated with standard visual field testing commonly available in local ophthalmology clinics.
"Educating patients and sharing the results with them can stimulate their willingness to comply with long-term follow-up visits and assessments." – Laura Omoboni, Executive Director of Clinical Trial Management, Medpace
Did you know that 28% of regulatory questions from authorities focus on study protocols? Families need clear explanations of these protocols to understand their impact on loved ones. RareLabs addresses this by offering plain-language updates and milestone reporting as part of their personalized drug discovery programs. This ensures families stay informed and engaged throughout the treatment journey.
Clear reporting is just the start - direct communication channels can further close any gaps in understanding.
Direct Access to Science Teams
As treatments move through rigorous testing, having uninterrupted access to science teams is critical. Open communication helps resolve issues quickly and ensures studies are designed with patient needs in mind. Advisory groups, for instance, allow patient advocates to provide direct input to research sponsors.
"Your protocol basically sets the scene for your label and the future use of your products in clinical practice." – Dr. Marco Tangelder, MD, PhD, Senior Medical Director, Medpace
During pivotal moments like drug administration day, families need immediate support. RareLabs offers direct access to their science team, enabling families to ask questions and address concerns without unnecessary delays. This kind of engagement helps clarify how outcome measures - whether primary or secondary - translate to real-world improvements in quality of life and functional abilities.
7. Prepare for Clinical Translation and Patient Access
Moving a treatment from the lab into patient care involves careful planning to meet FDA approval standards and ensure patients can access the therapy. On February 23, 2026, the FDA introduced draft guidance for individualized therapies aimed at genetic conditions. This guidance includes a "plausible mechanism" framework, which allows approvals based on solid scientific reasoning rather than relying solely on large-scale trials. RareLabs uses this framework to develop treatments for ultra-rare conditions where traditional trial methods aren’t feasible.
Regulatory and Post-Trial Considerations
Early collaboration with regulatory bodies is essential for navigating the approval process. Sponsors are encouraged to schedule pre-IND (investigational new drug) meetings to discuss trial designs and evidence requirements before starting key studies. This is especially important for therapies targeting very small patient populations - typically fewer than 1,000 individuals in the U.S. The FDA’s Rare Disease Evidence Principles (RDEP) process allows approval based on a single well-controlled study, paired with additional evidence from natural history data or mechanistic models.
"Going forward, the FDA's default position is that one adequate and well-controlled study, combined with confirmatory evidence, will serve as the basis of marketing authorization." – Martin A. Makary, FDA Commissioner
Even after approval, long-term monitoring remains vital. For gene therapies or other one-time treatments, the FDA generally requires 15 years of follow-up to track delayed adverse effects and confirm lasting benefits. Sponsors must plan for this extended monitoring, including securing funding and addressing challenges like transitions from pediatric to adult care.
RareLabs simplifies this process for families by incorporating clinical translation planning into its personalized drug discovery programs. They assist with identifying meaningful clinical endpoints and preparing for long-term regulatory responsibilities.
This foundational work ensures precision medicine can effectively demonstrate both target engagement and treatment success.
Integration with Precision Medicine
Precision medicine plays a key role in meeting regulatory requirements by providing targeted evidence. Under the plausible mechanism framework, pharmacodynamic biomarkers are essential to confirm target engagement - proving that the treatment has reached and altered its intended molecular target. For instance, in gene editing, showing that the genetic defect has been corrected at a molecular level is often more critical than visible clinical improvements.
Natural history studies add another layer of support by establishing baseline data for disease-specific biomarkers and outcomes. These studies can also act as external controls when using placebo groups is not ethical. Between 2016 and 2023, nearly 42% of approved gene therapies relied on natural history data or external controls instead of concurrent placebo groups.
The FDA’s growing acceptance of single pivotal trials backed by confirmatory evidence highlights the increasing reliance on precision medicine data. By 2025, about 50% of approved drugs were based on just one Phase 3 trial. This underscores the importance of robust data on target engagement and mechanistic proof to secure not only regulatory approval but also reimbursement from payers.
Conclusion: Summary of Key Considerations
Treating rare genetic diseases requires a patient-centered strategy that tackles unique regulatory and clinical hurdles. The seven-step checklist offers a clear guide for navigating the intricate process of personalized drug discovery. This includes evaluating therapeutic options, developing patient-specific cell models, ensuring safety validation, and preparing for clinical application.
Between January 2016 and September 2023, 12 gene therapies received approval - 42% of which utilized natural history data or external controls. These approvals highlight the importance of creative trial designs and meaningful regulatory collaboration.
The FDA has recently introduced initiatives aimed at accelerating rare disease drug development. These efforts are shaping current personalized treatment programs, offering families a clearer path from genetic profiling to clinical translation.
FAQs
How do I choose between gene therapy, ASOs, and small molecules?
Choosing the right approach - gene therapy, antisense oligonucleotides (ASOs), or small molecules - depends on the specific rare disease, its genetic basis, and the desired treatment outcomes.
- Gene therapy aims to address the underlying genetic issue, often designed as a one-time intervention.
- ASOs provide precise control by altering gene expression, though they require ongoing doses.
- Small molecules focus on managing symptoms or targeting biological pathways and are often taken orally, making them a flexible choice for treating a range of conditions.
Should we start with WES or WGS for genetic testing?
Starting with WES (whole exome sequencing) is a practical choice because it zeroes in on coding regions, where the majority of disease-causing mutations are found. This targeted approach makes it both cost-efficient and effective as an initial step. If WES doesn’t reveal the genetic cause, moving to WGS (whole genome sequencing) offers a broader analysis, covering non-coding regions and more intricate variations that WES might miss.
What proof does the FDA need to approve an ultra-rare treatment?
The FDA mandates substantial evidence to confirm both the effectiveness and safety of treatments for ultra-rare diseases. Given the limited number of patients available for study, traditional randomized controlled trials may not always be feasible. In such cases, the FDA allows the use of alternative data sources and tailored clinical trial designs. This approach aims to uphold strict standards for treatment approval while addressing the unique hurdles posed by ultra-rare conditions.



