Complete Guide to Personalized Medicine for Rare Diseases
Gene editing, ASOs and patient‑derived iPSCs are reshaping rare disease care, enabling faster, tailored treatments and adaptive regulatory pathways.

Personalized medicine is changing how rare diseases are diagnosed and treated. Instead of generic treatments, it uses genetic insights to target the root causes of conditions. Here's what you need to know:
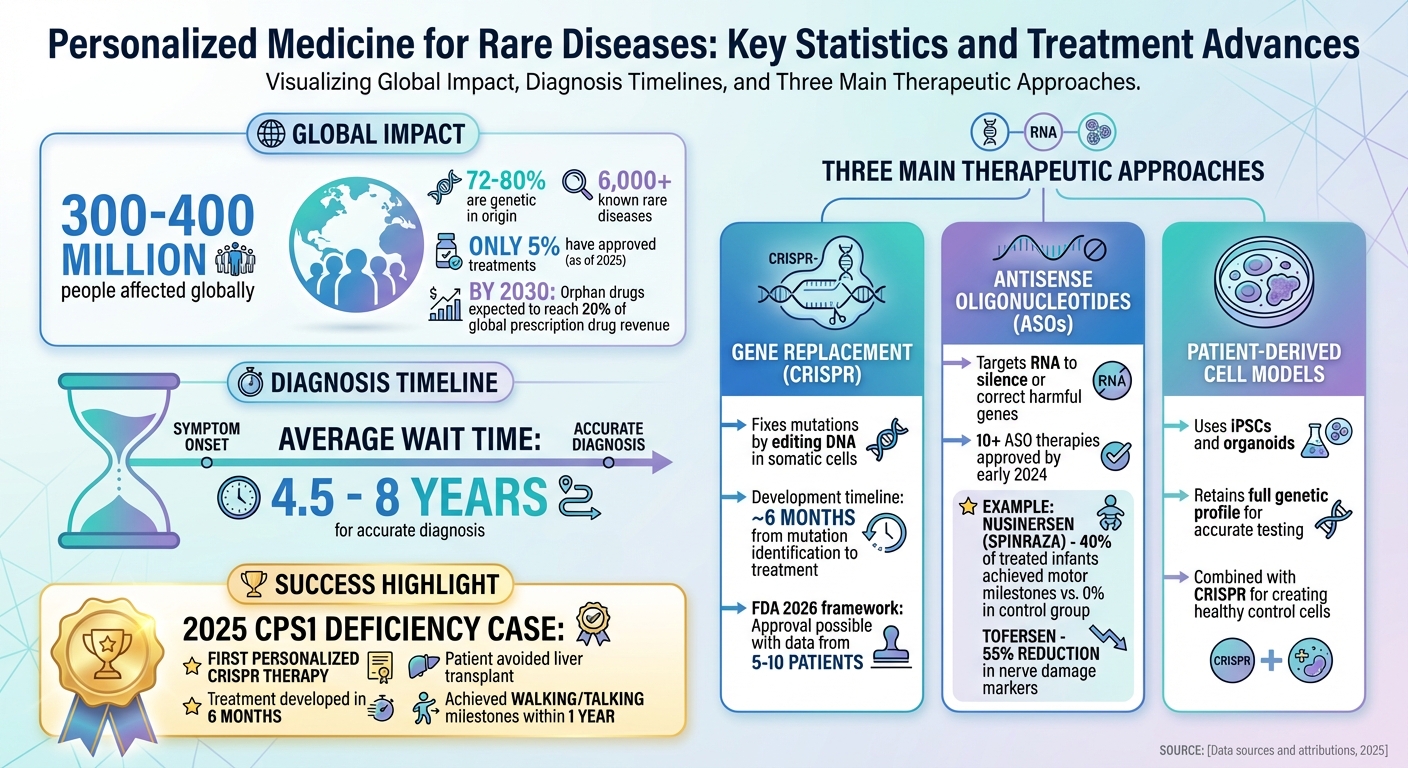
- Rare Diseases by the Numbers: Affect 300–400 million people globally; 72–80% are genetic. Only 5% of over 6,000 known rare diseases had approved treatments as of 2025.
- Diagnosis Challenges: Patients often wait 4.5–8 years for an accurate diagnosis.
- Therapy Advances:
- Gene Replacement: Fixes mutations using CRISPR to edit DNA in somatic cells.
- Antisense Oligonucleotides (ASOs): Targets RNA to silence or correct harmful genes.
- Patient-Derived Cell Models: iPSCs and organoids replicate patient-specific biology for testing treatments.
- Success Stories: In 2025, a CRISPR-based therapy helped an infant with CPS1 deficiency avoid a liver transplant, showing significant health improvements within a year.
- Regulatory Changes: The FDA's 2026 framework allows approval of therapies based on smaller studies, speeding up treatment availability.
Personalized medicine is addressing gaps in rare disease care, offering tailored solutions to patients who previously had no options. Keep reading to explore how these therapies work and their impact on healthcare.
Personalized Medicine for Rare Diseases: Key Statistics and Treatment Advances
Advancing Rare Disease Breakthroughs at Mayo Clinic
How Gene Replacement Therapies Work
Gene replacement therapies leverage CRISPR technology to edit DNA with pinpoint accuracy, addressing the mutations that cause rare diseases at their root. These therapies focus on somatic cells - non-reproductive cells found in organs such as the liver - ensuring that the genetic modifications affect only the individual receiving the treatment.
The process starts by identifying the specific mutation responsible for the disease in the patient’s DNA. Scientists then create a gene-editing tool to correct the mutation, typically within about six months. This tool is packaged into lipid nanoparticles, which act as protective carriers, safely delivering the genetic material through the bloodstream to the targeted organ.
Once administered, the lipid nanoparticles seek out the affected cells. Inside these cells, the gene editor makes precise changes to the faulty DNA, restoring the production of enzymes or proteins that were previously missing or defective. For metabolic disorders, this can mean the difference between severe complications and normal development.
"As a platform, gene editing -- built on reusable components and rapid customization -- promises a new era of precision medicine for hundreds of rare diseases, bringing life-changing therapies to patients when timing matters most: Early, fast, and tailored to the individual."
– Joni L. Rutter, Ph.D., Director of NIH's National Center for Advancing Translational Sciences (NCATS)
This platform-based strategy is reshaping how rare diseases are treated. Instead of creating entirely new drugs for each genetic mutation, researchers are reusing adaptable components to address various genetic variants. Under a new FDA framework introduced in February 2026, data from as few as 5 to 10 patients could be enough to approve an entire gene-editing platform.
The precision of this approach is setting a powerful example, as shown in the following case study.
Case Study: CPS1 Deficiency at Children's Hospital of Philadelphia
In February 2025, a six-month-old infant named KJ became the first person to receive a personalized CRISPR-based therapy for carbamoyl phosphate synthetase 1 (CPS1) deficiency, a rare metabolic disorder. CPS1 deficiency disrupts the breakdown of ammonia, leading to toxic buildups that can severely harm the brain and liver. Without intervention, a liver transplant would have been KJ’s only option.
Dr. Rebecca Ahrens-Nicklas from the Children’s Hospital of Philadelphia (CHOP) and Dr. Kiran Musunuru from Penn Medicine developed a tailored treatment within six months of identifying KJ’s genetic mutation. Using a base editor, they altered a single "letter" in the DNA sequence of KJ's liver cells, enabling the production of the CPS1 enzyme needed to convert toxic ammonia into urea.
Between February and April 2025, KJ received three infusions of the therapy. By February 2026, one year after treatment, he had reached key developmental milestones like walking and talking, could eat more dietary protein, and required less nitrogen-scavenging medication.
"While this treatment isn't a cure, after three infusions from February through April 2025, KJ has tolerated it well with no serious side effects. He's able to handle more dietary protein, requires less nitrogen‐scavenging medication, and we're seeing better control of ammonia levels during colds."
– Rebecca Ahrens-Nicklas, MD, PhD, Director of CHOP's Gene Therapy for Inherited Metabolic Disorders Frontier Program
CHOP is currently advancing cell and gene therapy research through 45 active pediatric clinical trials, supported by over 80 faculty members. KJ’s case highlights how personalized gene editing can provide transformative options for patients with few alternatives.
Antisense Oligonucleotide (ASO) Treatments for Genetic Mutations
Gene replacement therapies focus on correcting errors at the DNA level, but antisense oligonucleotides (ASOs) take a different route - targeting RNA. These synthetic, single-stranded nucleic acids (15–30 nucleotides in length) are designed to bind to specific mRNA sequences. Instead of introducing a new gene, ASOs intercept RNA instructions before they’re translated into proteins, either silencing harmful genes or correcting how they are read. This RNA-based method provides a complementary approach to traditional genome editing.
ASOs function in three main ways. The first is RNase H-dependent degradation, where the ASO binds to mRNA, triggering RNase H enzymes to destroy the mRNA and halt harmful protein production. The second mechanism, splice modulation (often called exon skipping), involves ASOs binding to pre-mRNA to bypass mutated exons, potentially restoring proper protein function. Lastly, ASOs can physically block mRNA, preventing it from being translated or interacting with proteins.
One of the standout features of ASO development is its relatively short timeline compared to traditional protein-targeted drugs. Since ASOs are designed based on the mRNA sequence rather than complex protein structures, the process from research to clinical application is more streamlined. This efficiency was highlighted by the development of Milasen, an ASO created specifically for a single patient with Batten disease, showcasing the potential for ultra-personalized treatments.
Several ASO therapies have already received FDA approval, offering new hope for rare diseases. For example, Nusinersen (Spinraza), approved in 2016 for spinal muscular atrophy (SMA), allowed 40% of treated infants to achieve motor milestones, compared to none in the control group. In Duchenne muscular dystrophy, Viltolarsen (approved in 2020) and Casimersen (approved in 2021) use exon skipping to address specific genetic mutations. More recently, Tofersen, targeting SOD1-related ALS, demonstrated a 55% reduction in plasma neurofilament light chain - a key marker of nerve damage.
"Since ASO design relies principally upon knowledge of mRNA sequence, the bench to bedside pipeline for ASOs is expedient compared with protein-directed drugs." – BioDrugs
Advancements in ASO chemistry, such as modifications to the sugar-phosphate backbone and bases, have improved their stability and resistance to degradation by enzymes. New delivery systems, including exosomes and specialized nanoparticles, are also expanding ASO reach to hard-to-target tissues like muscles. By early 2024, more than 10 ASO-based therapies had received regulatory approval for rare conditions.
Patient-Derived Cell Models in Drug Discovery
ASOs and gene therapies hold potential, but their success hinges on reliable testing systems. This is where patient-derived cell models come into play. These models are considered the gold standard for personalized treatments because they retain the full genetic profile of the patient. Unlike traditional animal studies or generic cell lines - which often miss the unique molecular features of rare genetic diseases - patient-derived models provide a more accurate platform for understanding and addressing these conditions. Alongside gene replacement and ASO therapies, these models deliver critical insights for developing targeted treatments.
The foundation of this approach lies in induced pluripotent stem cells (iPSCs), which are created by reprogramming a patient's somatic cells. These iPSCs act as a renewable, disease-specific cell source that can differentiate into relevant cell types or even form 3D organoids. These organoids replicate the structure and cellular complexity of native tissues, offering a more realistic model for studying diseases.
"The convergence of CRISPR genome editing, patient-derived organoids, and induced pluripotent stem cells (iPSCs) has reshaped in vitro disease modeling by enabling mechanistic investigations of human pathophysiology within genetically matched, tissue-relevant systems." – Stem Cell Research & Therapy
One of the key advantages of this system is the ability to use CRISPR technology to correct specific mutations in a patient’s disease-carrying cells. This process generates "healthy" cells that are genetically identical to the diseased ones, except for the corrected mutation. By comparing these CRISPR-corrected cells to the original diseased cells, researchers can confirm whether a particular mutation is responsible for the observed issues, ruling out the influence of unrelated genetic factors. For example, in February 2026, Lieberman et al. applied this approach to study NGLY1 deficiency. They created midbrain organoids from patient-derived iPSCs, which revealed reduced neuroepithelial expansion and mitochondrial dysfunction. These issues were successfully reversed in the CRISPR-corrected models, providing clear evidence of the mutation’s role in the disease.
RareLabs builds on these advancements by leveraging patient-derived models to push the boundaries of personalized drug discovery.
RareLabs' iPSC Creation and CRISPR-Corrected Controls
RareLabs incorporates patient-derived iPSC creation and CRISPR-corrected controls into its Personalized Drug Discovery Program. The process begins with reprogramming a patient’s cells into iPSCs, followed by thorough karyotyping quality assurance to ensure genetic stability. These iPSCs are then differentiated into disease-relevant cell types or assembled into 3D organoids that replicate the patient’s tissue environment. Using CRISPR genome editing, RareLabs corrects the specific mutation to create isogenic controls. These controls serve as precise comparators, enabling researchers to test multiple therapeutic strategies and identify the most effective, patient-specific treatment options.
RareLabs' Personalized Drug Discovery Program
RareLabs takes a cutting-edge approach to drug discovery by transforming patient-derived cell models into tailored treatments. By pinpointing whether a genetic defect occurs at the DNA, RNA, or protein level, they design therapies that directly address each patient's unique condition. Here's how their structured, step-by-step process works.
3-Step Approach: Modality Assessment, Screening, and Updates
The process begins with Modality Assessment, where advanced techniques like NGS (Next-Generation Sequencing) and WGS (Whole Genome Sequencing) identify whether the disease is monogenic, polygenic, or chromosomal. This classification helps determine the most suitable therapeutic strategy. For instance, a single-gene defect might benefit from gene replacement, while protein dysfunction could call for small molecule treatments.
The next step is Multi-Modality Screening, which leverages AI-driven tools like virtual screening, de novo molecule generation, and molecular docking to identify and refine potential treatment candidates. Patient-derived iPSCs (induced pluripotent stem cells) and CRISPR-corrected controls provide a realistic testing environment, enabling researchers to evaluate multiple therapies in conditions that closely mimic the patient's biology.
Finally, RareLabs ensures transparency and engagement through progress updates. Patients and caregivers receive milestone reports in plain language and have direct access to the science team for questions and insights.
Therapeutic Modalities: Small Molecules, ASOs, Gene Replacement, and Combinations
Once a candidate therapy is identified, RareLabs categorizes potential treatments based on the biological hierarchy of the defect. Here's how they approach different levels:
- Gene Replacement: Targets DNA-level defects by replacing faulty genes with functional copies.
- RNA-Based Therapies: Includes antisense oligonucleotides (ASOs), siRNA, and mRNA therapies to silence harmful transcripts or fix splicing errors at the RNA level.
- Small Molecule Drugs: Focuses on protein-level issues by altering enzyme activity or reshaping protein structures.
- Gene Editing (CRISPR/Cas9): Directly corrects genetic mutations at their source.
RareLabs follows a logical hierarchy for therapeutic selection: DNA-level interventions for structural abnormalities, RNA therapies for transcript-related issues, and protein-level solutions for functional problems. In some cases, combination therapies are explored, addressing multiple aspects of the disease simultaneously. These strategies are rigorously tested using patient-derived models, increasing the chances of finding effective treatments.
Challenges and Regulatory Progress in Rare Disease Therapeutics
Creating personalized treatments for rare diseases comes with a unique set of challenges that often prevent these therapies from reaching patients. For conditions that impact only a handful of individuals worldwide, traditional randomized controlled trials (RCTs) simply don’t work. Take carbamoyl-phosphate synthetase 1 (CPS1) deficiency, for instance - it affects just 1 in 1.3 million people. Similarly, fibrodysplasia ossificans progressiva has fewer than 1,000 known cases globally. Beyond the clinical difficulties, there’s little financial incentive for large pharmaceutical companies to invest, as the small patient populations rarely provide a meaningful return on investment.
"Add regulatory uncertainty, high per-patient development costs and uneven access to diagnostic testing and expert clinical care - it is clear why promising science often stalls before reaching patients."
– Dr. Rebecca Ahrens-Nicklas, attending physician at Children's Hospital of Philadelphia
Another major hurdle lies in manufacturing. While government funding often supports the early stages of research, the responsibility for producing these highly specialized therapies frequently falls on industry partners. These companies are forced to manufacture treatments "at risk", without any guarantee of reimbursement. As Sadik Kassim points out, industry players are motivated more by clear reimbursement pathways than by academic recognition. This creates a "last mile" problem, where scientific discoveries fail to evolve into real-world treatments due to gaps in manufacturing infrastructure and misaligned financial incentives. These challenges have pushed regulators to rethink how therapies for rare diseases are approved.
Regulatory frameworks are now starting to align with scientific advancements. On February 23, 2026, the FDA introduced draft guidance for its "Plausible Mechanism Framework." This framework focuses on therapies for genetic conditions with clear biological causes, allowing approval based on one well-controlled study supported by additional evidence.
"For ultra-rare conditions, randomized control trials are often - and mostly always - just not feasible. Under the framework that we're announcing today, one well-controlled clinical investigation, supported by confirmatory evidence, can support approval."
– Robert F. Kennedy Jr., Secretary of Health and Human Services
The framework takes a modular approach, enabling a single technology platform to address multiple mutations within the same gene - like updating a GPS with a new destination. It also allows for natural history data from untreated patients to serve as external controls and encourages the use of AI models and organs-on-chips to strengthen safety and efficacy data. These changes aim to lower regulatory barriers while still upholding safety standards, paving the way for more personalized therapies.
"It is our priority to remove barriers and exercise regulatory flexibility to encourage scientific advances and deliver more cures and meaningful treatments for patients suffering from rare diseases."
– Marty Makary, FDA Commissioner
A recent example highlights how these regulatory updates and manufacturing innovations can make a difference. In February 2025, a personalized CRISPR-based treatment for CPS1 deficiency was developed, produced, and administered within just six months. This achievement was made possible through a collaboration involving Children’s Hospital of Philadelphia, Danaher Corporation, and other partners. This case underscores how combining cutting-edge technologies with adaptive regulatory pathways can rapidly bring life-saving treatments to patients who previously had no options.
Future Directions in Personalized Medicine for Rare Diseases
The treatment landscape for rare diseases is undergoing a transformation as platform technologies replace the traditional one-drug-one-disease model. Instead of crafting separate treatments for every genetic variant, researchers are now creating modular systems capable of addressing multiple mutations within the same gene. Think of it like software updates - these platforms can be reconfigured to suit different patients’ needs. This shift is paving the way for new approaches that could reshape the industry entirely.
A major milestone in this evolution was the launch of Aurora Therapeutics in February 2026. Co-founded by Nobel laureate Jennifer Doudna and geneticist Fyodor Urnov, the company was designed to utilize the FDA's new plausible mechanism pathway. This pathway enables the development of genome-editing treatments for ultra-rare diseases that were once considered too costly to tackle. Aurora’s approach marks a significant change in how biotech companies are addressing rare disease drug development.
As these adaptable platforms gain traction, advancements in artificial intelligence (AI) and next-generation sequencing are enhancing early diagnosis and personalized treatment. AI and machine learning are speeding up discovery by sifting through massive genomic datasets, helping to detect rare diseases faster and predict which treatments will work best for individual patients. Paired with modular gene therapy platforms, these tools are refining precision medicine. Additionally, next-generation sequencing techniques like whole-genome sequencing (WGS) and whole-exome sequencing (WES) allow doctors to pinpoint single-gene mutations with incredible accuracy. This means interventions can now happen earlier - before irreversible damage occurs.
Economic trends are also aligning with these scientific breakthroughs. By 2030, orphan drugs are expected to account for nearly 20% of global prescription drug revenue. To encourage investment in ultra-rare disease therapies, the FDA is working to reduce development costs and regulatory hurdles. As FDA Commissioner Marty Makary explained:
"We don't want investors scared away because of the high cost. We want investors to see the bright promise of rare disease therapies".
On a global scale, the World Health Assembly's 2025 resolution aims to establish an international framework to improve access for underserved patients. This initiative promotes collaboration and data-sharing, a crucial step since 95% of rare diseases still lack approved therapies. The goal is clear: ensure that no one is left without a diagnosis or treatment simply because their condition is rare.
Conclusion
Personalized medicine is reshaping how rare diseases are treated. Techniques like gene replacement therapy, antisense oligonucleotides, and CRISPR gene editing are now targeting the underlying genetic causes of these conditions.
Even though millions of patients around the world still lack approved treatments, the field is advancing quickly. Recent developments highlight how customized therapies can provide real clinical benefits for patients who previously had no options. As FDA Commissioner Marty Makary aptly put it, "For patients and families, there is no time to wait".
Since about 80% of rare diseases have genetic origins, genomic research plays a key role in treatment. Tools like whole-genome sequencing can pinpoint harmful mutations within days - or even hours - of birth, opening the door to early interventions that could prevent irreversible damage.
RareLabs' personalized drug discovery program is leading by example. By combining advanced diagnostics with customized therapeutic approaches, the program uses patient-derived iPSCs, CRISPR-corrected controls, and a range of therapeutic options - from small molecules to gene replacement therapy. Families are kept in the loop with plain-language updates and direct access to the science team, ensuring transparency throughout the process.
The treatment landscape for rare diseases is evolving. With regulatory frameworks adapting to match scientific progress and new technologies emerging, RareLabs is dedicated to ensuring that no patient is overlooked. Together, these advancements are paving the way for a future where every rare disease patient has access to a treatment tailored to their unique genetic profile.
FAQs
Am I eligible for a personalized gene-editing or ASO treatment?
Eligibility for treatment often hinges on several factors, including the specific genetic disorder, your unique genetic makeup, and the therapies currently available. In most cases, genetic testing is necessary to confirm the diagnosis and identify the mutation involved. Specialists will evaluate whether your condition meets the criteria for treatments, which could include participation in clinical trials or access through compassionate use programs. It's essential to consult a healthcare provider with expertise in rare genetic conditions to explore your options fully.
What genetic test should I ask for to speed up diagnosis?
To diagnose rare genetic diseases more efficiently, consider comprehensive genetic testing options such as targeted gene panels, exome sequencing, or genome sequencing. These tests can pinpoint disease-causing variants across numerous genes. Newer techniques like long-read genome sequencing and optical genome mapping are also proving useful for identifying complex genetic changes. Consulting with a genetic specialist can help you determine the best testing approach based on your symptoms and family history.
How do patient-derived iPSCs or organoids help pick the best therapy?
Patient-derived induced pluripotent stem cells (iPSCs) and organoids offer a way to replicate a patient’s condition in the lab using their own cells. These models closely resemble the affected tissues, allowing researchers to dive deep into understanding how diseases work. Even better, clinicians can use these models to test treatments directly, helping to pinpoint the most effective options for each individual. This method is reshaping how rare diseases are approached, making care more precise and personalized.



