Ultimate Guide to Orphan Drug Designation Process
Clear guide to FDA orphan drug designation: eligibility, 90-day review, tax credits, fee waivers, and 7-year market exclusivity.

Orphan Drug Designation (ODD) helps pharmaceutical companies develop treatments for rare diseases affecting fewer than 200,000 people in the U.S. by offering financial and regulatory incentives. These include tax credits, a $4.5 million FDA fee waiver, and 7 years of market exclusivity after approval.
Key Takeaways:
- Eligibility: The drug must target a rare condition or an orphan subset and show potential effectiveness.
- Application Process: Requires detailed documentation, including disease prevalence and scientific evidence.
- FDA Review: Takes 90 days; outcomes include approval, requests for more data, or denial.
- Benefits:
- 25% tax credit for clinical trials in the U.S.
- Waiver of FDA application fees.
- Access to grants and priority review vouchers worth $75M–$150M.
- Market exclusivity for 7 years, protecting against competition for the same indication.
This program makes rare disease treatments financially viable and accelerates drug development using advanced research approaches like patient-specific cell models.
FDA Orphan Drugs Program and Financial Incentives for CDER Medical Products- June 10, 2019
Eligibility Requirements for Orphan Drug Designation
To meet the criteria, your drug must address a rare disease and provide credible evidence of its potential effectiveness. The FDA assesses this based on three key factors: the rarity of the disease, the scientific foundation supporting your drug, and whether your treatment offers benefits over existing options.
What Qualifies as a Rare Disease
A rare disease is defined as one affecting fewer than 200,000 individuals in the United States. However, there’s an alternative scenario: even if a condition impacts more than 200,000 people, it may still qualify if U.S. sales are unlikely to recoup research and development costs.
Prevalence estimates must be current at the time of application. When multiple estimates are available, the FDA typically accepts the highest figure unless you provide a valid reason to use a lower one. Use updated U.S. Census data to back your prevalence claims. If relying on foreign data, you’ll need to explain its relevance to the U.S. population.
Another option is through orphan subsets. If your drug is only suitable for a specific group within a broader disease population due to its properties, you can apply for a subset designation. For instance, a drug might only be effective for patients with a specific genetic mutation within a larger disease category. These prevalence guidelines set the foundation for the next step: providing scientific evidence.
Required Scientific Evidence
Your application must include a medically sound basis to show the drug’s potential effectiveness for the rare disease. This can be supported by clinical trials, preclinical studies using relevant animal models, or strong in vitro research that outlines a clear mechanism of action.
"In order to designate a product as an orphan drug, the scientific rationale portion of the designation application must include enough information to establish a medically plausible basis for expecting the drug to be effective in the rare disease." - FDA
If human data are unavailable, preclinical animal studies can suffice. In cases where neither human data nor a suitable animal model is available, the FDA may consider alternative forms of evidence, such as insights into the disease’s biology, your drug’s mechanism of action for that condition, and laboratory studies. However, animal toxicology data, which focuses on safety rather than effectiveness, will not fulfill the scientific rationale requirement.
It’s critical to provide all relevant data, whether the findings are positive, negative, or inconclusive. Include full copies of referenced materials, not just abstracts, and for online sources, submit a hard copy along with the URL and access date. These rigorous standards ensure the drug has the potential to meaningfully improve treatment options.
Advantages Over Current Treatments
If your drug is considered similar to an already approved treatment, you must demonstrate clinical superiority to gain designation. The FDA assesses this based on greater effectiveness, improved safety for a significant number of patients, or a notable contribution to patient care.
For designation, you only need to propose a plausible hypothesis of clinical superiority. However, to secure market exclusivity after approval, you must prove that superiority, often through direct clinical comparisons. Greater effectiveness might involve better outcomes on biomarkers or clinical endpoints, while improved safety could mean reduced side effects or fewer interactions with other drugs.
A major contribution to patient care might include practical improvements like shorter treatment durations, less discomfort, simpler administration methods (e.g., oral instead of IV), longer intervals between doses, or the potential for self-administration. However, the FDA generally doesn’t consider cost or compliance improvements unless they directly enhance safety or effectiveness.
How to Apply for Orphan Drug Designation
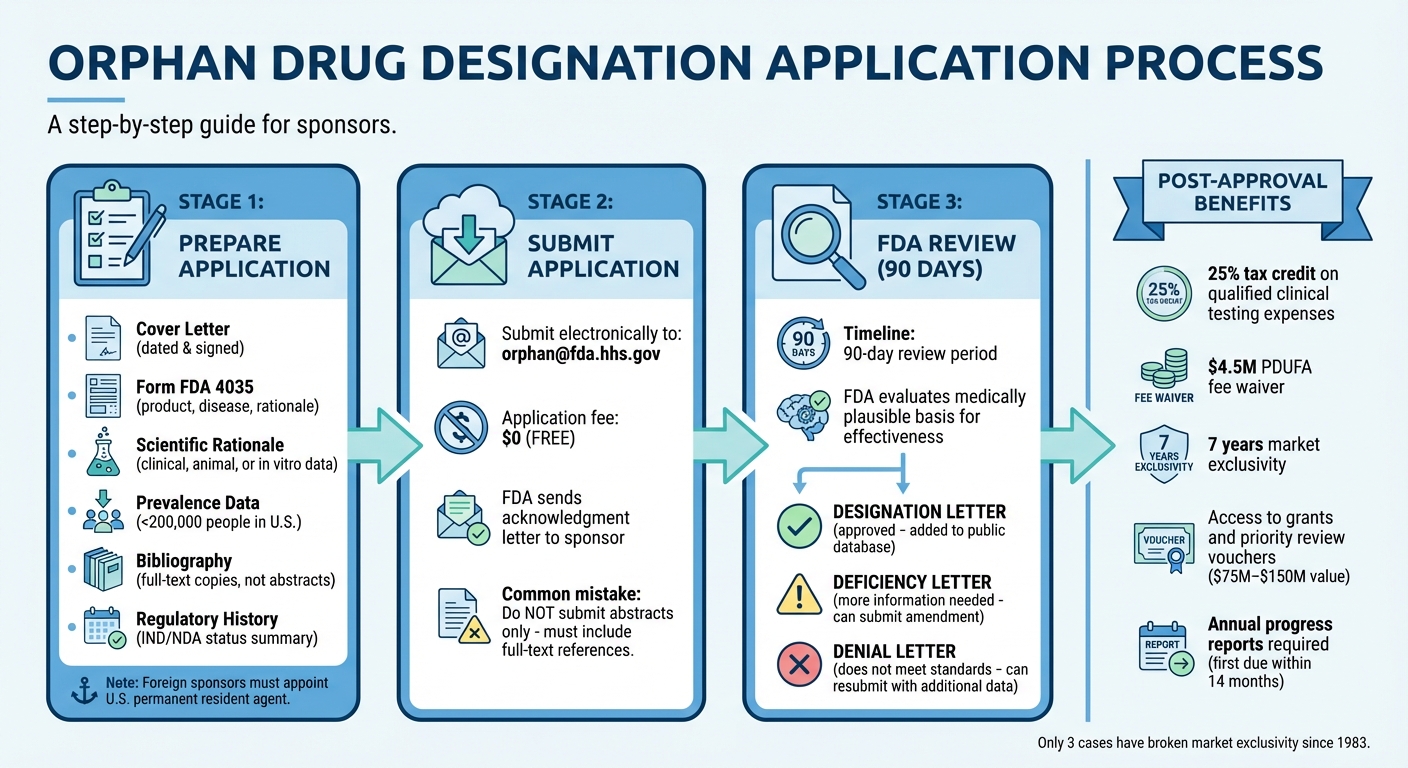
Orphan Drug Designation Application Process and Benefits Timeline
This section walks you through the key steps to turn a promising treatment into an FDA-designated orphan drug. From preparing documents to submitting your application and navigating the review process, here's what you need to know.
Preparing Your Application Documents
Your application must include several critical components: administrative details, a thorough drug description, a clear explanation of the disease, prevalence data, and a strong scientific rationale. Start with a cover letter that’s dated and signed by a sponsor representative. Use Form FDA 4035 to ensure all fields required under 21 CFR 316.20 are addressed.
The drug description should provide detailed information about the active moiety (for small molecules) or the primary molecular features (for macromolecules), along with its physical and chemical properties. For the disease description, identify the rare condition, outline its pathophysiology, explain the intended use of your drug, and justify its necessity.
When it comes to prevalence data, include authoritative references, detailed calculations, and full-text copies of all sources - not just abstracts. Use the latest U.S. Census Bureau data to back up your claims. If your drug has been marketed internationally, summarize its regulatory status, including any IND or NDA approvals.
For foreign sponsors, appointing a U.S. permanent resident agent is mandatory. This agent will file the application and act as the primary contact.
| Required Document | Document Requirements |
|---|---|
| Cover Letter | Must be dated and signed by a sponsor representative |
| Form FDA 4035 | Includes product details, disease description, and scientific rationale |
| Scientific Rationale | Supported by clinical, animal, or in vitro data showing efficacy |
| Prevalence Data | Proof that fewer than 200,000 people in the U.S. are affected |
| Bibliography | Full-text copies of all references and dated website printouts |
| Regulatory History | Summary of IND/NDA status and any global regulatory actions |
Once you've gathered and organized your documents, you're ready to submit your application.
Submitting Your Application
Submit your completed application electronically to orphan@fda.hhs.gov. There’s no fee for submitting an orphan drug designation application. After submission, the FDA will send an acknowledgment letter to the sponsor or their appointed agent.
A common mistake to avoid: submitting only abstracts instead of full-text references. The FDA does not accept abstracts.
FDA Review Timeline and Outcomes
The FDA typically reviews applications within 90 days, coordinating with other agencies to confirm eligibility. During this review, the agency evaluates whether the application establishes a medically plausible basis for the drug's expected effectiveness. This determination relies on supporting evidence such as clinical data, preclinical animal models, or in vitro studies.
"The FDA can come back during the 90‑day review with an information request that the sponsor needs to address to avoid the rejection of the application." - Halloran Consulting Group
At the end of the review, there are three possible outcomes:
- Designation Letter: Issued if the drug meets all criteria.
- Deficiency Letter: Requests additional information or data.
- Denial Letter: Issued if the application does not meet the necessary standards.
If you receive a deficiency or denial letter, you can address the feedback, gather the required data, and submit an amendment to your application.
Once the orphan drug designation is granted, the FDA adds the sponsor name, drug name, and designated orphan use to its public searchable database. However, details about rejections or withdrawals are not made public.
What You Gain from Orphan Drug Designation
Securing orphan drug designation offers a mix of financial incentives and regulatory support that can help offset the high costs of developing treatments for rare diseases. These benefits make what might seem like a risky endeavor into a more feasible and appealing opportunity.
Tax Credits and Fee Waivers
One major advantage is the Orphan Drug Tax Credit (ODTC), which allows you to claim a federal tax credit equal to 25% of qualified clinical testing expenses. However, this credit only applies to clinical trials conducted after receiving the designation, and preclinical research or animal testing costs don't count. To qualify, trials must take place in the U.S. or its territories. Claiming the credit requires filing IRS Form 8820 ("Orphan Drug Credit") and reporting it on Form 3800 ("General Business Credit"). Eligible expenses include payments to trial participants, test administration, patient monitoring, and investigator compensation. If the credit exceeds your tax liability for the year, you can carry it back one year or forward for up to 20 years. Keep in mind, though, that you can't claim the same expenses under both the ODTC and the Research & Experimentation (R&E) Tax Credit - they must be allocated to one or the other.
In addition to tax credits, sponsors are exempt from Prescription Drug User Fee Act (PDUFA) application fees, which can save millions in regulatory costs. You may also qualify for federal grants through the FDA's Orphan Products Grants Program to support clinical trials.
7-Year Market Exclusivity
Once your orphan drug is approved by the FDA, you gain seven years of market exclusivity. This means the FDA cannot approve another version of the same drug for the same use or indication during this time.
"Orphan exclusivity isn't a patent. It's a separate legal shield." - RxDeal
Breaking this exclusivity is extremely rare. Since the Orphan Drug Act was enacted in 1983, only three cases have succeeded in meeting the FDA’s clinical superiority standard. To do so, competitors must demonstrate improved effectiveness, better safety, or a significant contribution to patient care, such as easier administration or reduced treatment burden.
A recent clarification under the Consolidated Appropriations Act, effective February 3, 2026, limits exclusivity strictly to the approved use or indication, rather than the entire orphan-designated disease. This means other companies can still develop treatments for different indications within the same rare disease.
For drugs targeting rare pediatric diseases, there's an added perk: a Priority Review Voucher (PRV). These vouchers allow expedited FDA review for a future product and can be sold on the secondary market for $100 million to $200 million. The FDA is authorized to award PRVs through September 30, 2029.
FDA Support During Development
The FDA provides extensive guidance throughout the development process. This includes protocol advice on clinical trial design to ensure data collection aligns with regulatory standards for rare disease populations. If your drug is similar to an already approved orphan product, the FDA offers a path to designation and exclusivity if you can show clinical superiority, such as easier administration or reduced treatment burden.
Foreign sponsors also benefit from administrative support, including a U.S.-based permanent resident agent required to handle correspondence and annual reporting with the FDA. This ongoing support helps streamline the development process and align your efforts with regulatory requirements, making it easier to navigate the challenges of rare disease drug development.
Combining Orphan Drug Designation with Personalized Drug Discovery
For ultra-rare genetic diseases, combining orphan drug designation with personalized drug discovery simplifies the development process. This strategy aligns with one of the FDA's key requirements: providing a "medically plausible basis" to expect a drug to be effective. By tailoring treatments to individual genetic profiles, researchers can build a strong foundation for advanced experimental models.
Using Patient-Derived Cell Models
To meet the FDA's need for a medically plausible basis, in vitro patient-specific models play a critical role. These models offer essential evidence, especially when suitable animal models are unavailable or fail to mimic human disease accurately. The FDA accepts data from patient-derived cell lines to support orphan drug designation applications.
RareLabs specializes in creating patient-derived iPSC models to evaluate drug efficacy against specific genetic variants. These models include CRISPR-corrected controls and karyotyping to ensure precision.
"In the absence of human data and when a relevant animal model does not exist, the FDA may consider a combination of alternative data that includes... supporting in vitro data." - FDA
A noteworthy example comes from the UCLA Care and Research in Neurogenetics (CARING) Clinic. Between 2019 and 2024, this clinic used genomics-first methods to assess 316 patients with neurodevelopmental disorders. Pathogenic or likely pathogenic variants were identified in 152 patients, and personalized findings led to immediate clinical management changes for 36 of them. This underscores how personalized data strengthens orphan drug applications by providing clear scientific rationale.
Multiple Treatment Approaches
RareLabs employs a variety of therapeutic strategies that align with the advantages of orphan drug designation. Historically, small molecules have been a dominant choice, accounting for 77.6% of orphan FDA approvals between 1983 and 2007. Other promising approaches include antisense oligonucleotides (ASOs) and gene replacement therapies, both of which fit within the FDA's "Plausible Mechanism Framework." This framework allows for market approval based on targeted, small-scale clinical studies for ultra-rare conditions.
When single treatment methods prove insufficient, RareLabs develops combination strategies. Each approach undergoes a Personalized Modality Assessment, ensuring it is tailored to the specific genetic disease. These diverse options are supported by a robust tracking system that ensures steady progress and compliance with regulatory requirements.
Tracking Progress and Milestones
Once orphan drug designation is granted, the FDA requires annual progress reports outlining preclinical and clinical milestones. The first report is due within 14 months of designation. RareLabs assists with this process by providing clear, plain-language updates and milestone tracking throughout the drug development journey. Their science team remains accessible, ensuring all stakeholders stay informed and that documentation meets regulatory standards. This structured approach helps keep the entire project on track, from discovery to treatment development.
Conclusion
The orphan drug designation program offers key financial incentives and regulatory benefits that make developing treatments for rare diseases more manageable. These include tax credits covering 25% of qualified clinical testing expenses, a waiver of the hefty $4.5 million Prescription Drug User Fee Act (PDUFA) fee, and 7 years of market exclusivity. Together, these measures help offset the challenges of creating therapies for conditions that affect fewer than 200,000 Americans. This framework also encourages the use of advanced research techniques during drug development.
Incorporating patient-specific iPSC models into the designation process not only aligns with FDA evidence requirements but also highlights the importance of personalized, early-stage therapeutic strategies. These patient-derived cell models can strengthen designation applications, especially when conventional research methods fall short of providing sufficient data.
"Supporting the development and evaluation of new treatments for rare diseases is a key priority for the FDA." - FDA
Applying for orphan drug designation early - when patient-derived models or preliminary data suggest potential efficacy - can significantly enhance a program's value. Early designation opens doors to crucial regulatory support, which can be instrumental throughout the drug development process. With over 7,000 rare diseases affecting approximately 300 million people globally, the demand for novel treatment solutions continues to rise.
FAQs
When should we apply for orphan drug designation?
The best time to seek orphan drug designation is before kicking off clinical trials. Why? Early applications help sponsors take full advantage of perks like tax credits, user fee waivers, and market exclusivity once the drug gets approved. While the FDA does accept applications at any point in development, submitting early is often the smartest move. The agency reviews these requests by examining factors such as the disease’s progression, expected outcomes, and how resistant it is to treatment - all in relation to the proposed drug.
How do we prove U.S. disease prevalence under 200,000?
The FDA classifies a rare disease as one that impacts fewer than 200,000 individuals in the United States. To establish this, credible evidence is necessary. This could include epidemiological studies, patient registries, or other trustworthy sources that clearly show the disease's prevalence falls within this limit.
What happens if the FDA sends a deficiency or denial letter?
If the FDA issues a deficiency letter regarding an orphan drug designation request, it indicates that the request might not fully satisfy the requirements for designation. To assist sponsors in addressing these issues, the FDA offers guidance aimed at resolving deficiencies and facilitating a smoother review process. Although the exact steps to take after receiving a deficiency or denial letter aren’t explicitly outlined, the FDA does provide a clear framework for submitting and reviewing designation requests.



