No Treatment Options: How Families Find New Solutions
Families facing rare genetic diseases fund research, build registries, use iPSCs and custom therapies when no approved treatment exists.
When faced with rare genetic diseases, families often discover that 95% of rare diseases lack FDA-approved treatments. This forces them to take matters into their own hands by funding research, collaborating with scientists, and even creating personalized treatments. With over 30 million Americans affected by rare diseases - and half of these being children - the stakes are high. Families are leveraging advanced tools like genetic sequencing, patient-derived cell models, and therapies such as gene replacement or antisense oligonucleotides to address these challenges.
Key insights:
- Diagnosis delay: It can take over 5 years to diagnose a rare disease.
- Economic hurdles: Drug companies often avoid rare diseases due to low profitability.
- Personalized medicine: Families drive solutions, raising millions to fund research and treatments.
- New tools: Technologies like iPSCs and AI-powered patient registries are accelerating progress.
This article explains how families are navigating these challenges, the tools they use, and the personalized therapies they pursue to create hope where standard treatments fall short.
Philadelphia doctors use gene editing therapy to treat child with rare disease for the first time
How Families Are Leading the Search for Treatments
When pharmaceutical companies step away from rare diseases, families often step in to fill the void. They're not just waiting for solutions - they're actively funding research, creating infrastructure, and collaborating with scientists to push treatments forward. As Tania Simoncelli, Vice President of Science in Society at the Chan Zuckerberg Initiative, puts it:
"The patient community has to do that work. They shouldn't have to, but they have to."
Families are no longer just participants in the process - they're becoming leaders. By launching nonprofits, forming global patient networks, and even starting their own biotech companies, they’re ensuring that drug development remains focused and driven. They’re also learning the technical aspects of drug discovery, negotiating with experts, and addressing key bottlenecks, such as manufacturing viral vectors or developing cell models, that might otherwise delay progress. For many, this journey begins right after diagnosis, as they rally resources to fund targeted research.
What Happens After Diagnosis
The first step following a diagnosis is often genetic confirmation. Families turn to whole exome or genome sequencing to identify the specific mutation responsible for the disease. Once they have this information, they dive into advocacy - reading scientific studies, reaching out to researchers worldwide, and gaining a deep understanding of the disease mechanisms.
Take Ryan and Jenny Bragg, for example. After their daughter Clara was diagnosed with GM1 gangliosidosis, they raised over $1 million in under a year to support research at Auburn University. Their efforts directly funded the $750,000 needed to manufacture a viral vector for a clinical trial - work that would have been delayed by 18 months if reliant on NIH grants. As Jenny Bragg explained:
"We're actually trying to fund the cure, not find the cure. We've already found it."
Many families also join specialized training programs to better navigate the complex drug development process. Initiatives like the RARE Bootcamp from the EveryLife Foundation and RareLaunch from the National Organization for Rare Disorders equip parents with the tools they need to make informed decisions and drive progress.
Creating Patient Registries
Patient registries play a vital role in advancing research. These databases collect natural history data - tracking symptoms, disease progression, and outcomes - which helps attract researchers and meets regulatory requirements for clinical trials.
The Cure Mito Foundation offers a powerful example. This parent-led nonprofit launched the Leigh Syndrome Global Patient Registry in September 2021. By June 2025, it included 402 participants from 48 countries. This registry has already supported the development of multiple gene therapy candidates and repurposed drugs now entering early trials.
In late 2020, Cure Mito also gathered 40 skin biopsies from patients across nine countries for an FDA Investigational New Drug (IND) application. Nasha Fitter, co-founder of the FOXG1 Research Foundation, summed up the challenges families face:
"It's insane, as parents, that we're building mouse models and cell lines. It's insane to me that this is what we have to do."
Modern registries are using technology to make the process easier for families. Platforms now utilize FHIR (Fast Healthcare Interoperability Resources) APIs to automatically pull structured data - like labs, vitals, and medications - from hospital systems. AI tools can extract information from unstructured clinical notes, while mobile surveys allow families to report outcomes directly. To ensure the data meets regulatory standards, registries adopt global frameworks like CDISC and the OMOP Common Data Model, making it ready for FDA or EMA submissions.
High-quality registry data can even become a source of funding. Pharmaceutical companies often license this longitudinal data for clinical trial recruitment or use it as synthetic control arms, creating a sustainable model for ongoing research.
Using Patient-Derived Cell Models for Drug Discovery
After families receive genetic confirmation of their child's diagnosis, the next step often involves creating patient-derived cell models. These models allow researchers to study the disease in a controlled lab environment and test potential treatments before they are ever administered to the patient. One of the most effective tools for this research is induced pluripotent stem cells (iPSCs) - cells that are reprogrammed from a patient’s skin or blood into any type of cell relevant to the disease.
What sets patient-derived iPSCs apart from engineered lab models is their ability to capture the entire genetic profile of the individual. This includes not only the mutation causing the disease but also other genetic factors that can influence the disease's progression and the patient's response to treatments. This level of precision is especially important for rare diseases, where genetic differences can lead to vastly different outcomes. By using these models, researchers can bridge the gap between diagnosis and actionable treatment strategies.
How iPSCs Are Made from Patient Cells
The process begins with a simple sample, such as skin cells from a biopsy or blood cells. Scientists then reprogram these cells into a pluripotent state, meaning they can develop into any type of cell in the body. From there, the iPSCs are differentiated into the specific cell types affected by the disease - neurons for neurological conditions or liver cells for metabolic disorders, for example.
To ensure these cells are accurate, researchers use methods like karyotyping and long-range PCR. They also create CRISPR-corrected control lines, which allow them to compare diseased cells with healthy ones. This comparison confirms that observed changes are directly caused by the mutation, not by other genetic factors. By leveraging these precise models, researchers can test therapies tailored to the patient’s unique genetic makeup more efficiently.
Advancements in technology have significantly sped up this process. Where it once took months to derive corrected iPSCs from patient fibroblasts, newer protocols can now achieve this in just four weeks. This faster timeline is critical for families dealing with diseases that progress rapidly.
Examples of iPSC-Based Treatments
These methods have already reshaped how treatments are developed. One groundbreaking example is Milasen, a treatment created for Mila Makovec, who had CLN7 Batten disease. In 2017, Dr. Timothy Yu and his team at Boston Children’s Hospital identified a unique mutation in Mila’s CLN7 gene using whole-genome sequencing. They then created iPSCs from Mila’s cells to test custom antisense oligonucleotides (ASOs) in the lab, ultimately finding the most effective candidate.
By early 2018, within just one year, the FDA approved a single-patient clinical trial, and Mila received her first dose. The results were remarkable: her seizures decreased from 30 per day to between 5 and 12 per day, and their duration dropped from about two minutes to just a few seconds. Julia Vitarello, Mila’s mother and founder of Mila’s Miracle Foundation, shared:
"Batten is really cruel, and she lost a lot before she began therapy. But to me it seems her disease has absolutely stopped."
Dr. Yu likened this approach to a surgical intervention rather than a traditional medication:
"In a way, this is more like a bone marrow transplant or a surgery than a pill."
Mila’s case was a milestone for N-of-1 therapeutics - treatments designed specifically for a single patient based on their unique genetic profile. While making these treatments scalable remains a challenge, her story shows how patient-derived cell models can speed up drug discovery, cutting timelines from years to months and offering hope to families facing rare and devastating diseases.
Different Types of Personalized Therapies
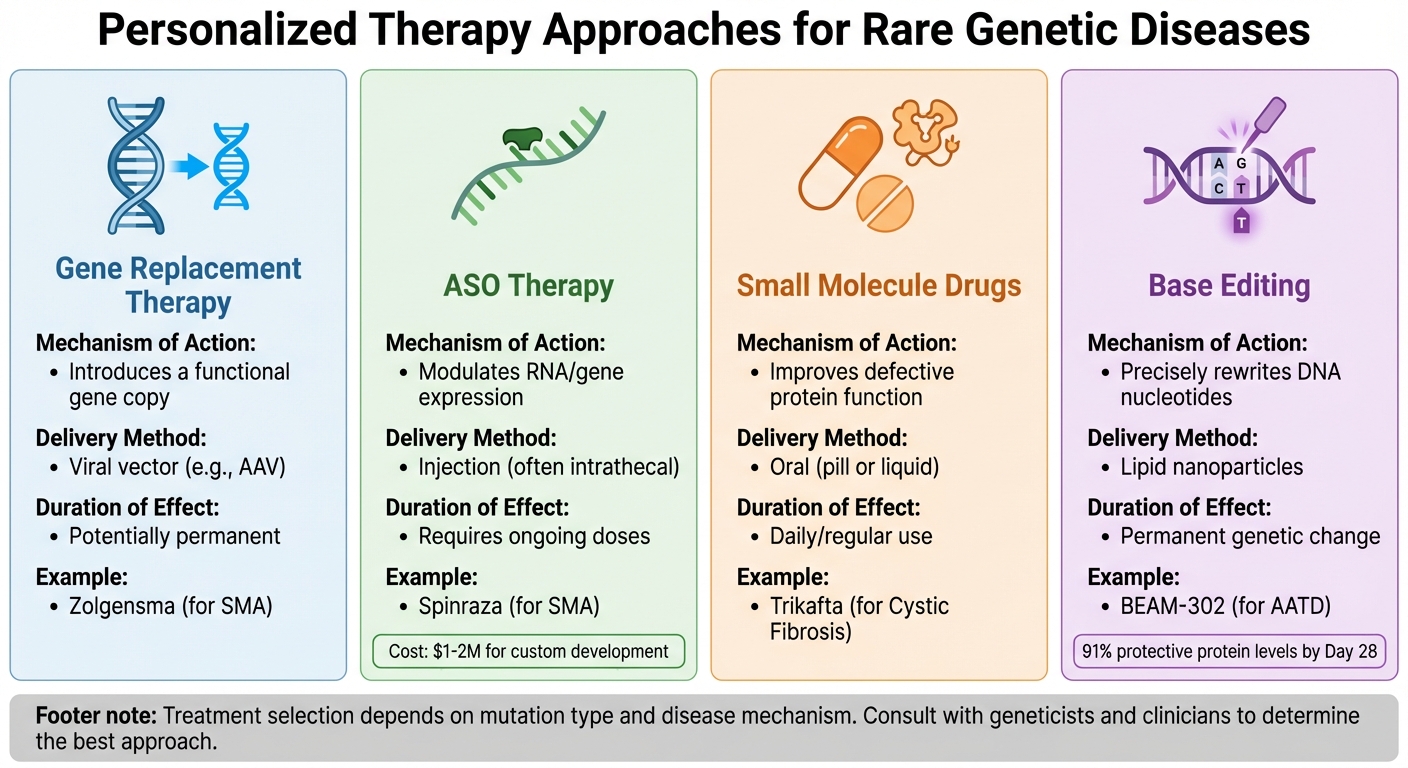
Comparison of Personalized Therapy Types for Rare Genetic Diseases
Thanks to patient-derived models, families now have a clearer path to explore targeted therapies. These models help identify the most effective treatments based on the specific mutation, its impact on protein function, and the disease's cellular mechanisms. Researchers often focus on four main therapeutic approaches for rare genetic diseases, each with distinct methods and applications.
Gene Replacement Therapy involves replacing a faulty gene with a functional copy using a viral vector, like AAV, in a single intervention. A well-known example is Zolgensma, used for spinal muscular atrophy (SMA).
Antisense Oligonucleotide (ASO) Therapy utilizes synthetic nucleotide strands to bind specific RNA sequences. This process adjusts gene expression by modifying protein production or correcting splicing errors without altering the DNA itself. Unlike gene replacement, ASO therapies require ongoing injections, often intrathecal. For instance, Spinraza, a treatment for SMA, is administered regularly and comes with high costs. Developing a custom ASO for an individual patient can cost between $1 million and $2 million.
Small Molecule Drugs focus on improving the function of defective proteins produced by mutated genes. These drugs are typically taken orally, either as a pill or liquid. Trikafta, a treatment for cystic fibrosis, is a prime example of this approach.
Base Editing takes precision to the next level by rewriting individual DNA nucleotides (e.g., changing adenine to guanine) without causing double-strand breaks. In a 2025 Phase 1/2 clinical trial, Beam Therapeutics demonstrated the potential of this approach with BEAM-302. A single 60 mg dose corrected the PiZZ genotype in alpha-1 antitrypsin deficiency patients, increasing protective M-AAT protein levels to 91% of total circulating AAT by Day 28.
Comparing Treatment Approaches
| Therapy Type | Mechanism of Action | Delivery Method | Duration of Effect | Example |
|---|---|---|---|---|
| Gene Replacement | Introduces a functional gene copy | Viral vector (e.g., AAV) | Potentially permanent | Zolgensma (for SMA) |
| ASO Therapy | Modulates RNA/gene expression | Injection (often intrathecal) | Requires ongoing doses | Spinraza (for SMA) |
| Small Molecule | Improves defective protein function | Oral (pill or liquid) | Daily/regular use | Trikafta (for Cystic Fibrosis) |
| Base Editing | Precisely rewrites DNA nucleotides | Lipid nanoparticles | Permanent genetic change | BEAM-302 (for AATD) |
Selecting the right therapy depends on understanding the mutation's specific impact. For example, gene replacement is ideal when a gene is entirely nonfunctional, while small molecule drugs can enhance the performance of a flawed protein. ASO therapy is particularly useful for addressing issues like splicing errors. These tailored approaches highlight the importance of working closely with experts to align treatments with a patient's unique genetic profile.
Working with Scientists and Clinicians
Once the best treatment approach is identified, collaboration with geneticists, clinicians, and bioinformaticians becomes crucial. After pinpointing the mutation, specialists review possible therapies to determine the best match. For example, they might assess whether a proven ASO platform can be customized for the patient’s specific mutation. Patient-derived induced pluripotent stem cells (iPSCs) are also invaluable for testing how a drug reacts to the patient’s genetic background.
To speed up the development of personalized treatments, the FDA introduced the "Plausible Mechanism Pathway" in November 2025. This initiative fast-tracks therapies based on evidence from a small number of cases. FDA Commissioner Marty Makary emphasized:
"The FDA will work as a partner and guide in ushering these therapies to market, and our regulatory strategies will evolve to match the pace of scientific advances."
This new pathway is already having an impact. In early 2025, Baby K.J., a newborn diagnosed with a severe metabolic disorder, received a custom gene-editing therapy within a week of diagnosis. The treatment, approved under the Plausible Mechanism Pathway, led to a 50% reduction in the need for ammonia-clearing medication and improved protein processing.
Families are encouraged to join patient registries and collaborate with specialized research institutions like Boston Children's Hospital or nonprofits such as the n-Lorem Foundation, which develops personalized ASOs for nano-rare patients at no cost. Communities like KIF1A.org can also help families identify existing ASOs relevant to their child’s mutation.
The bottom line: precision is key. While over 90% of rare diseases currently lack approved treatments, the combination of detailed genetic data, expert teams, and tailored therapies is helping families move from diagnosis to actionable solutions faster than ever before.
Staying Informed Throughout the Process
Developing personalized therapies is no quick endeavor - it’s a journey that can span months or even years. Regular updates are essential, as they provide clarity at every stage, from creating patient-specific cell models to screening drugs and, potentially, clinical testing. Without consistent communication, uncertainty only grows deeper.
Understanding the science behind the process helps parents make informed decisions about treatment risks. Danny Miller, a parent of children with MEPAN syndrome, shared his experience of navigating the early, uncertain days:
"It was very scary... As a parent, you go through a lot of self-doubt, a lot of blame. You wonder, 'What did we do wrong?' ... We were both really determined to try to find answers, and we weren't getting them."
Having direct access to researchers and receiving transparent progress reports can significantly reduce that uncertainty.
But it’s not just about peace of mind; staying engaged also helps keep the research moving forward. Families who participate in regular scientific meetings, ask questions, and even contribute to hypothesis development often become active collaborators in the research process. This ongoing dialogue helps bridge the gap between lab discoveries and the decisions families need to make.
What to Expect from RareLabs
For families leading drug discovery efforts, staying informed is essential. RareLabs ensures that updates are clear, concise, and easy to understand. Instead of overwhelming technical jargon, families receive milestone summaries that explain key discoveries, their relevance to the condition, and the next steps in the process. This way, even parents without a scientific background can stay engaged and ask meaningful questions.
RareLabs also prioritizes direct communication. Beyond standard email updates, families have real-time access to the scientists and clinicians working on their program. You can schedule calls to discuss findings, clarify confusing data, or share observations about your child’s symptoms that might shape the research. Communication isn’t an afterthought - it’s built into the process from the very beginning.
This level of transparency and accessibility ensures families remain active participants in the journey, laying the groundwork for meaningful involvement in the research process.
How to Stay Involved
There are several ways families can remain engaged and contribute to the advancement of personalized therapies:
- Join or establish a patient registry: Patient-owned registries, like the INSIGHTS registry, allow families to share valuable data that can accelerate research for others with the same diagnosis. These registries also help researchers identify patterns across patients, which can speed up drug discovery.
- Collaborate through open-science platforms: Networks like the N=1 Collaborative enable families to share data, methods, and insights. These platforms create opportunities to pool resources and avoid duplicating efforts, saving both time and money.
- Advocate for policy changes: By joining a Rare Disease Advisory Council (RDAC) or signing up for action alerts from organizations like the EveryLife Foundation, families can influence public policy and stay informed about regulatory developments. These actions can have a direct impact on treatment timelines and the progress of personalized medicine.
Staying informed and involved isn’t just empowering - it’s a critical part of driving the development of treatments tailored to rare conditions.
Results from Families Who Pursued Personalized Medicine
Treatment Advances for Rare Diseases
In January 2018, Mila Makovec became the first person to receive a drug tailored specifically to her genetic mutation. Mila, diagnosed with Batten disease, endured up to 30 seizures a day. Dr. Timothy Yu at Boston Children's Hospital used whole-genome sequencing to uncover an error in her MFSD8 gene and created an antisense oligonucleotide called milasen to target it. Unlike traditional drugs that often take over a decade to develop, this treatment was ready in just 12 months.
The results were remarkable. After starting milasen, Mila’s seizures nearly stopped. She also regained the ability to eat without needing a feeding tube. The development of milasen was funded by $3 million raised through Mila's Miracle Foundation. Mila’s mother, Julia Vitarello, who founded the foundation, reflected on the immense effort:
"Creating Mila's drug was like climbing Everest without oxygen or guides. But we need a clear path and to lower Everest."
Another inspiring case is Rich Horgan, who sought a solution for his brother Terry, diagnosed with an ultra-rare form of muscular dystrophy. Horgan founded Cure Rare Disease, an organization dedicated to developing custom gene therapies. By collaborating with biotech companies and raising funds through philanthropy, the organization has since expanded its mission to create personalized therapies for other families facing similar challenges. These stories highlight how families can play a pivotal role in advancing science when no standard treatments are available.
These achievements also serve as a foundation for RareLabs' personalized approach to rare disease treatments.
How RareLabs Customizes Programs for Each Family
RareLabs builds on the successes of family-driven breakthroughs by offering personalized therapy programs tailored to each family's specific needs. Using advanced iPSC (induced pluripotent stem cell) technology, RareLabs develops customized therapeutic strategies, which may include small molecule screening, antisense oligonucleotide therapy, gene replacement, or even a combination of these approaches.
Costs and timelines vary depending on the patient's condition and the complexity of the program. Families are kept informed every step of the way with clear, straightforward updates at key milestones. RareLabs also ensures direct communication with the scientific team working on the case, allowing families to ask questions, share observations about symptoms, and stay actively involved in the process. This open, collaborative approach ensures families remain integral to the journey toward a treatment.
Conclusion: How Families Can Take Action
When faced with a rare disease that lacks an existing treatment, families can take proactive steps to make a difference. The journey begins with securing a precise genetic diagnosis through Whole Genome Sequencing. This advanced testing can identify elusive mutations that standard methods might overlook. As shown by advancements like customized iPSC therapies, having an accurate diagnosis can pave the way for targeted research and help overcome funding and regulatory challenges. From there, families can collaborate with experts to create patient-specific cell models and explore therapies tailored to their child’s unique genetic mutation.
Navigating the complex regulatory landscape, such as applying for Single-Patient Compassionate-Use INDs, is another critical step. This process often requires significant financial resources - frequently exceeding $2 million. Julia Vitarello, founder of Mila's Miracle Foundation, captured the urgency and importance of these efforts:
"I feel like I owe it to the millions of children and families who will follow Mila to do whatever I can to keep the momentum going for individualized medicines"
Families can also connect with established patient registries and advocacy networks. Sharing data and insights through these channels not only helps drive research forward but also reduces costs associated with developing new treatments.
Organizations like RareLabs play a pivotal role by using patient-derived iPSC technology to create personalized therapies. Whether it’s small molecule screening, ASO therapy, or gene replacement, families are kept informed at every stage. RareLabs ensures transparent communication, providing updates and maintaining a close partnership with families throughout the process.
Stories like Mila Makovec’s demonstrate that taking immediate and personalized action can change lives. With more than 30 million people in the U.S. affected by rare diseases - and only 5% of these conditions having approved treatments - the need for decisive action is clear. For families ready to explore personalized medicine, RareLabs offers the expertise and resources to transform hope into meaningful progress.
FAQs
What are the first steps after a rare genetic diagnosis?
After receiving a rare genetic diagnosis, the first step is to fully understand the condition and tap into available resources. Work closely with healthcare providers and researchers to make sense of the diagnosis. They can help identify potential clinical trials or connect you with support groups that offer guidance and community.
Many families take action by fundraising for drug screening efforts or forming partnerships with biotech companies to push treatment development forward. Tools like patient-derived cell models and cutting-edge approaches, including gene replacement therapies or small molecule screening, are paving the way for more personalized treatment options. These efforts can bring hope and progress in addressing rare genetic conditions.
How do patient registries speed up treatment development?
Patient registries play a key role in speeding up treatment development for rare diseases. By gathering detailed genetic, clinical, and demographic data, these registries enable researchers to uncover patterns, track how diseases progress, and identify therapy targets more quickly. They also simplify the process of recruiting participants for clinical trials, offer real-world data to support personalized treatments, and encourage collaboration among families, researchers, and biotech companies. These combined efforts help cut down delays and make it possible to deliver new therapies to patients more efficiently.
How do families get FDA access for a single-patient therapy?
Families have the option to obtain single-patient therapies through the FDA's expanded access, or "compassionate use", program. To begin, a physician must submit a request for an Investigational New Drug (IND) application to the FDA and also receive approval from the drug manufacturer. After both approvals are granted, the FDA assigns an IND number. This number authorizes the drug to be shipped to the treating physician for use with the patient. The process involves providing thorough details about the patient and the proposed treatment.



