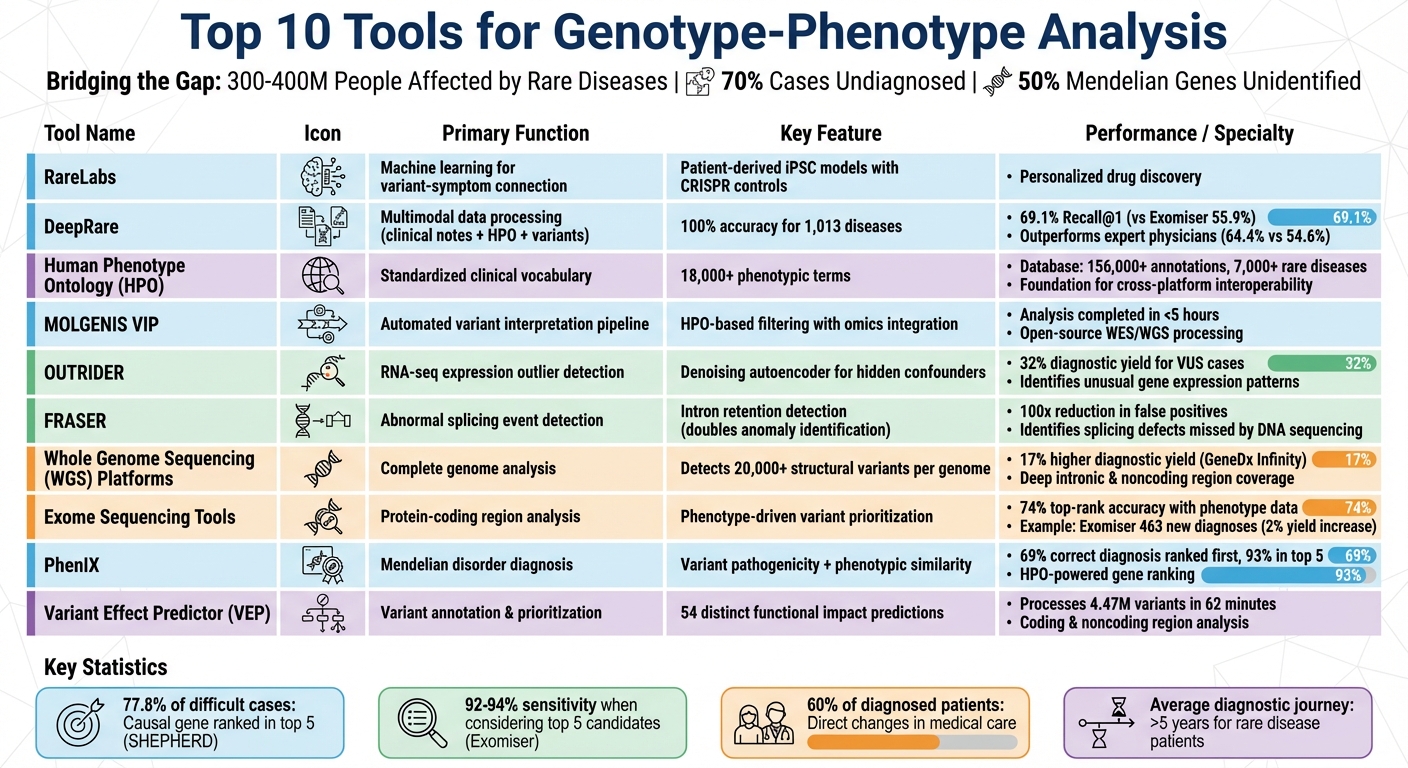
Top Tools for Genotype-Phenotype Analysis
Comprehensive roundup of 10 tools that link genetic variants and clinical phenotypes to speed rare disease diagnosis and personalize care.
Genotype-phenotype analysis is key to diagnosing rare diseases, with 300–400 million people affected globally. Despite advancements in sequencing technologies, 70% of cases remain undiagnosed, and 50% of Mendelian genes are still unidentified. This gap highlights the importance of advanced computational tools that integrate genetic and clinical data to streamline diagnostics and support personalized treatments.
Here are 10 standout tools for genotype-phenotype analysis:
- RareLabs: Uses machine learning to connect genetic variants with clinical symptoms, offering tailored diagnostics and therapeutic insights.
- DeepRare: Processes multimodal data with high accuracy, outperforming experts in rare disease diagnosis.
- Human Phenotype Ontology (HPO): Standardizes clinical symptoms into computable formats, aiding in precise genomic comparisons.
- MOLGENIS VIP: An open-source pipeline automating variant interpretation with HPO-based filtering.
- OUTRIDER: Identifies unusual gene expression patterns in RNA-seq data, addressing variants of uncertain significance.
- FRASER: Detects abnormal splicing events in RNA-seq data, focusing on rare splicing anomalies.
- Whole Genome Sequencing (WGS) Platforms: Provide comprehensive genomic data, identifying deep intronic and structural variants.
- Exome Sequencing Tools: Focus on protein-coding regions, integrating phenotype data for better diagnostic accuracy.
- PhenIX: Links genetic variants with clinical symptoms for Mendelian disorder diagnosis.
- Variant Effect Predictor (VEP): Analyzes and prioritizes genomic variants across coding and noncoding regions.
These tools leverage cutting-edge technologies to improve diagnostic precision, reduce manual effort, and connect genetic discoveries to actionable clinical insights. They enable researchers and clinicians to navigate complex rare disease cases with greater efficiency, ultimately supporting patient-specific care.
Top 10 Genotype-Phenotype Analysis Tools for Rare Disease Diagnosis
MPG Primer: Genotype, Phenotype, and GWAS Data (2026)
1. RareLabs
RareLabs focuses on a patient-first approach, combining machine learning with personalized drug discovery. Instead of sticking to generic diagnostic tools, it bridges the gap between identifying genetic variants and crafting customized therapeutic solutions for individuals with rare genetic disorders.
Integrating Genotype and Phenotype Data
RareLabs uses machine learning to merge VCF genotype data with HPO-encoded phenotype data. This process links clinical symptoms directly to genetic evidence, even when clinical data is variable.
The platform takes things further by employing statistical methods to pinpoint "comorbid" phenotypes - clinical features that tend to appear together more often than by chance. Through network analysis, these phenotypes are grouped into functional clusters, which are then tied to specific genes and molecular systems. This approach allows clinicians to see not only which genes are involved but also how they fit into the patient's overall clinical picture. This level of data integration brings clarity and precision to diagnostics.
Delivering Personalized Insights
RareLabs doesn't just stop at integrating data - it provides actionable, traceable diagnostics tailored to individual patients. By combining a patient’s unique HPO terms with biomedical knowledge graphs, the platform can identify causal genes, even for rare or previously unknown genetic conditions.
Once potential genes are identified, RareLabs employs a structured three-step process. This involves creating patient-derived iPSC models with CRISPR-corrected controls, which researchers can use to test various therapeutic approaches. These include small molecule screening, antisense oligonucleotide therapy, and gene replacement therapy, all customized to the patient’s genetic profile. This comprehensive method directly informs personalized treatments, making RareLabs a standout in rare disease research.
2. DeepRare
Relevance to Rare Disease Research
DeepRare is designed to tackle one of the biggest challenges in rare disease research: lengthy diagnostic timelines. By processing multimodal data - like free-text clinical notes, HPO (Human Phenotype Ontology) terms, and genetic variants - it significantly speeds up the process.
A study published in Nature in February 2026 analyzed nine datasets (6,401 cases involving 2,919 rare diseases). The results were striking: DeepRare achieved 100% diagnostic accuracy for 1,013 diseases, including those in the "long tail" of rare diseases that are often harder to identify. Its architecture is specifically designed to integrate genotype and phenotype data effectively, making it a powerful tool for rare disease diagnostics.
Ability to Integrate Genotype and Phenotype Data
DeepRare operates on a three-tier architecture that connects a central host, specialized agent servers (featuring over 40 tools), and external databases like PubMed and OMIM. The system processes patient data through two parallel branches:
- The phenotype branch standardizes HPO terms and pulls relevant literature.
- The genotype branch annotates and ranks genetic variants based on their clinical importance.
This dual-branch setup allows DeepRare to combine a patient’s specific multisystem symptoms with their genomic data. In multi-modal tests, it outperformed other tools, achieving a Recall@1 score of 69.1%, compared to Exomiser’s 55.9%. Users can submit raw VCF files and clinical notes, and the system automatically extracts standardized HPO terms for analysis.
Accuracy and Reliability of Results
DeepRare’s design translates into exceptional diagnostic accuracy. In a test involving five experienced rare disease physicians and 163 cases, DeepRare achieved a Recall@1 score of 64.4%, outperforming the experts’ 54.6%. Additionally, an independent review by ten associate chief physicians on 180 randomly selected cases confirmed a reference accuracy of 95.4%.
"This is one of the first demonstrations of a computational model surpassing expert physicians on rare disease phenotype-based diagnosis."
– Arkadi Mazin, Journalist, Lifespan.io
DeepRare also ensures transparency by providing traceable reasoning chains that link its diagnostic suggestions to supporting evidence.
Support for Personalized or Patient-Specific Insights
DeepRare doesn’t rely on one-size-fits-all solutions. If an initial diagnostic hypothesis doesn’t meet the required criteria, the system revisits earlier steps, gathering additional patient-specific evidence until a diagnosis is reached. This iterative process ensures diagnoses are tailored to the unique clinical presentation of each patient.
The system also ranks genetic variants by their clinical significance and generates clear reasoning chains. These chains connect diagnostic outputs to relevant clinical guidelines and research, ensuring users can trace the logic behind each suggestion. For local installations, the system requires at least 16GB RAM (32GB recommended), 100GB of storage, Java 21+, Python 3.8+, and Exomiser with its ~20GB dataset.
3. Human Phenotype Ontology (HPO)
Relevance to Rare Disease Research
The Human Phenotype Ontology (HPO) plays a key role in rare disease research by standardizing clinical data for better genotype-phenotype analysis. With a vocabulary of over 18,000 terms to describe phenotypic abnormalities, HPO provides a framework that supports accurate diagnosis of rare genetic conditions.
Currently, HPO includes more than 156,000 annotations related to hereditary diseases, with over 116,000 annotations for more than 7,000 rare diseases. In 2024, the HPO consortium expanded its resources by adding 2,239 new terms and 49,235 new annotations, working alongside experts in fields like psychiatry and cardiology. Major initiatives, such as the 100,000 Genomes Project in the UK and the NIH Undiagnosed Diseases Network in the U.S., rely on HPO for their research into rare diseases.
Ability to Integrate Genotype and Phenotype Data
HPO serves as a computational framework that bridges the gap between molecular biology and clinical disease by linking gene variants to their associated clinical features. Built as a directed acyclic graph, HPO uses logical definitions from resources like PATO and UBERON to enable computational tools to calculate semantic similarity. This structure also allows for cross-species phenotype comparisons, connecting human data with model organisms such as mice and zebrafish.
"The HPO can act as a scaffold for enabling the interoperability between molecular biology and human disease."
- PubMed
This interoperability is crucial for tools like Exomiser and Genomiser, which integrate phenotypic data with sequencing results to prioritize candidate genes associated with diseases. Additionally, HPO is a core element of the GA4GH Phenopacket Schema, which standardizes the exchange of phenotypic data across various platforms. This integration strengthens the diagnostic and analytical precision of downstream tools.
Support for Personalized or Patient-Specific Insights
HPO facilitates precision medicine by enabling deep phenotyping, which helps rank differential diagnoses based on a patient’s unique clinical features. Tools like Phenomizer apply semantic similarity algorithms to assess the "Information Content" of symptoms, narrowing down potential genetic disorders with greater specificity.
In testing, HPO-based concept recognition demonstrated a precision of 82.8% for immune system diseases and 77.8% for nervous system diseases. The HPO consortium is also working to embed the ontology and the GA4GH Phenopacket Schema into Electronic Health Records (EHRs), making rare disease data more accessible for personalized clinical care. To support global diagnostics, HPO is freely available and has been translated into 10 languages, including Chinese, Japanese, and Spanish. By standardizing clinical phenotypes, HPO equips diagnostic tools to provide more tailored and precise insights for patients.
4. MOLGENIS VIP
MOLGENIS VIP pushes the boundaries of rare disease diagnostics by offering an automated pipeline for variant interpretation. With its ability to integrate omics data and Human Phenotype Ontology (HPO)-based filtering, it simplifies the complex process of identifying genetic variants in rare disease cases.
Relevance to Rare Disease Research
MOLGENIS VIP, short for Variant Interpretation Pipeline, is an open-source tool that automates the annotation, filtering, and classification of genetic variants from Whole Exome Sequencing (WES) and Whole Genome Sequencing (WGS) data. It has become a cornerstone for international rare disease projects, including the European Rare Diseases Research Alliance (ERDERA) and Solve-RD.
In 2024, the EU project "Solve-RD" utilized MOLGENIS VIP to analyze data from 41 rare disease patients who had previously gone undiagnosed through standard clinical methods. By leveraging the pipeline's omics integration and HPO-based filtering, researchers successfully identified causal variants in cases that had remained unsolved. Additionally, the Society of Genome Diagnostics Laboratories (VKGL) in the Netherlands validated its performance by analyzing 25,664 classified variants and confirming diagnoses for 18 patients from routine clinical diagnostics.
Ability to Integrate Genotype and Phenotype Data
A standout feature of MOLGENIS VIP is its seamless integration of genetic data with clinical symptoms. By using HPO terms, the tool filters and prioritizes variants that align with a patient’s specific presentation. Since version 9.1.0, released in March 2025, default reports now include full HPO labels for enhanced clarity.
"VIP can process short- and long-read data from different platforms and offers tools for increased sensitivity: a configurable decision-tree and HPO-based, gene inheritance filters to pinpoint causal variants." - medRxiv Preprint
The pipeline also generates interactive, template-based reports, enabling clinical experts to interpret prioritized variants alongside phenotypic and gene inheritance data. This includes family-based analyses, such as identifying compound heterozygotes or variants with incomplete penetrance. These capabilities make it a powerful tool for refining diagnostic accuracy.
Accuracy and Reliability of Results
MOLGENIS VIP incorporates CAPICE, a gradient boosting model for predicting pathogenicity, which has demonstrated superior performance compared to other methods. Once set up, a diagnostic professional can complete a full analysis in under 5 hours.
The pipeline supports both short- and long-read sequencing data from platforms like Illumina, Oxford Nanopore, and PacBio. It also integrates third-party tools such as VEP, DeepVariant, and AnnotSV, ensuring a thorough and reliable analysis of genetic variants.
Support for Personalized or Patient-Specific Insights
MOLGENIS VIP excels at delivering patient-specific insights by automatically filtering variants based on detailed HPO terms. This functionality allows clinicians to tailor the analysis to each individual case.
"MOLGENIS VIP is a flexible human Variant Interpretation Pipeline for rare disease using state-of-the-art pathogenicity prediction (CAPICE) and template-based interactive reporting to facilitate decision support." - MOLGENIS
The system is highly adaptable, supporting custom Ensembl VEP plugins and various sequencing platforms. For mitochondrial genome analysis, it includes integrated scores like MitoTip and APOGEE, further expanding its diagnostic capabilities in recent versions.
5. OUTRIDER
OUTRIDER (OUTlier in RNA‐seq fInDER) is an R-based tool designed to identify unusually expressed genes in RNA sequencing data, particularly for diagnosing rare diseases. Using a denoising autoencoder approach, it models expected read counts while adjusting for hidden confounders and technical noise that could otherwise lead to false positives. This makes it especially valuable when DNA sequencing alone doesn't provide clear answers.
Relevance to Rare Disease Research
As part of the Detection of RNA Outliers Pipeline (DROP), OUTRIDER plays a pivotal role in interpreting Variants of Uncertain Significance (VUS) that DNA sequencing struggles to resolve. For example, in a 2025 study involving 34 patients with undiagnosed neurodevelopmental disorders, a multi-omics approach using OUTRIDER achieved a diagnostic yield of 32%, identifying an average of 4 outliers per patient.
"RNA sequencing (RNA‐seq) has emerged as a companion diagnostic tool that provides functional evidence to interpret VUS by detecting aberrant RNA phenotypes." - npj Genomic Medicine
Around 26% of VUS are thought to involve RNA phenotypes, such as frameshifts or splice disruptions, which OUTRIDER can help identify. By calculating P-values and Z-scores, it highlights outliers where read counts significantly deviate from expected patterns.
Ability to Integrate Genotype and Phenotype Data
While OUTRIDER focuses on transcriptomic data, it can also integrate with genomic data to uncover causal variants linked to observed expression changes. In the 2025 study, researchers used OUTRIDER to analyze participant SF197, a patient with an undiagnosed neurological disorder. The tool detected a significant reduction in GFM1 expression (fold change: 0.35, Z-score: -9.76, FDR: 4.46×10⁻¹⁵). This finding led to a closer look at exome data, revealing a 104.5 kb deletion that removed 11 enhancers for GFM1. This discovery ultimately provided a diagnosis, ending the patient’s diagnostic journey. Such cases highlight how transcriptomics can complement genomics in personalized diagnostics.
Support for Personalized or Patient-Specific Insights
OUTRIDER stands out for its ability to deliver insights tailored to individual patients by identifying unique expression outliers. For instance, in the same 2025 study, participant SF185 showed a 0.71 fold change and a Z-score of -4.13 for MSTO1. This prompted further investigation, which uncovered a heterozygous splice region variant (c.967-3C>A). This variant caused exon elongation and triggered nonsense-mediated decay, explaining the patient’s symptoms. These findings can guide personalized therapeutic strategies, such as drug repurposing or the development of antisense oligonucleotide therapies specific to the patient’s genetic profile.
6. FRASER
FRASER (Find RAre Splicing Events in RNA-seq) is a specialized computational tool designed to detect abnormal splicing events in RNA sequencing data. Unlike tools that focus on gene expression, FRASER zeroes in on splicing anomalies - a common but often overlooked factor in rare diseases. As part of the Detection of RNA Outlier Pipeline (DROP), FRASER provides a crucial layer of analysis by identifying splicing defects that standard DNA sequencing methods typically miss.
Relevance to Rare Disease Research
Splicing defects are a significant factor in many rare diseases, but they often go undetected by exome or genome sequencing. FRASER bridges this gap by identifying both alternative splicing and intron retention events, two mechanisms that can disrupt normal gene function. Notably, its ability to include intron retention detection can double the number of splicing anomalies identified compared to tools that ignore this factor.
"The FRASER framework and workflow aims to assist the diagnostics in the field of rare diseases where RNA-seq is performed to identify aberrant splicing defects." - gagneurlab, GitHub
FRASER has proven its value in clinical diagnostics by identifying cases that other tools missed. For example, it detected pathogenic intron retention events in the MCOLN1 gene and reprioritized exon truncations in the TAZ gene - key findings that other splicing tools failed to uncover.
Accuracy and Reliability of Results
FRASER employs a denoising autoencoder to account for hidden confounding factors in RNA-seq data, combined with a beta-binomial model to analyze read count ratios. This approach enables it to differentiate between genuine pathogenic events and background noise or common variations. This precision translates into a 100-fold reduction in false positives compared to standard z-score methods, all while maintaining high sensitivity.
The latest version, FRASER 2.0, introduces several advancements. It uses the Intron Jaccard Index as its primary splice metric, with a refined delta cutoff of 0.1 (lower than the previous 0.3). Additionally, the Optimal Hard Threshold (OHT) accelerates the algorithm's processing speed by 6–10×.
Integration of Genotype and Phenotype Data
FRASER enhances diagnostic workflows by focusing exclusively on splicing events. While it primarily analyzes transcriptomic data, it complements genomic sequencing by providing functional evidence of splicing disruptions. This is particularly useful when genomic data reveals variants of uncertain significance, as FRASER can confirm whether those variants affect splicing - offering critical insights into their pathogenicity.
Personalized Insights for Precision Medicine
FRASER generates patient-specific metrics like Z-scores, p-values, and delta-PSI (percent spliced in) to identify individual splicing anomalies. These insights are invaluable for developing precision therapies, such as antisense oligonucleotide (ASO) treatments, which can correct specific splicing errors. This capability makes FRASER a powerful tool for advancing personalized medicine in rare disease cases where standard treatments are unavailable.
7. Whole Genome Sequencing (WGS) Platforms
Moving beyond transcriptomic analyses, whole genome sequencing (WGS) platforms offer a full genomic snapshot, making them a powerful tool for diagnosing rare diseases.
WGS platforms examine the entire genome, including noncoding and deep intronic regions. They can detect a wide array of genetic variations such as small variants, copy number variants (CNVs), structural variants (SVs), mitochondrial variants, and repeat expansions.
Relevance to Rare Disease Research
WGS has shown great promise in diagnosing cases that remain unresolved after clinical exome testing. For example, the Undiagnosed Diseases Network conducted a study from 2016 to 2018 with 465 patients, finding that WGS provided unique diagnoses in 8% of previously unsolved cases. Additionally, WGS identified multilocus diagnoses in 5% of resolved cases.
Cutting-edge tools like the DRAGEN pipeline, which leverages graph references and machine learning, have improved the accuracy of variant detection. GeneDx’s "Infinity" dataset - comprising nearly 1 million exomes and genomes alongside 8 million phenotypic data points - achieved a diagnostic yield that was 17% higher than traditional methods. Moreover, approximately 60% of individuals who received a genomic diagnosis experienced direct changes in their medical care.
Accuracy and Reliability of Results
WGS platforms are now capable of detecting over 20,000 structural variants per genome. When combined with trio testing (sequencing a patient along with both biological parents), the chances of identifying causative genetic changes increase significantly, while reducing the number of uncertain variants. A study by Baylor Genetics in 2025/2026 highlighted the effectiveness of Emedgene’s Explainable AI (XAI), which achieved 97% accuracy in prioritizing genetic insights - rising to 98% for trio testing.
"Emedgene's machine learning simplifies the highly complex task of variant analysis, allowing us to handle more tests every day."
- Dr. Ray Louie, PhD, Assistant Director, Greenwood Genetic Center
The Greenwood Genetic Center adopted Emedgene’s AI platform, cutting their genomic analysis turnaround time by 75%. This efficiency allowed them to process a greater volume of tests daily. By streamlining workflows, AI-powered tools significantly enhance the speed and precision of genomic data analysis, paving the way for tailored treatment approaches.
Support for Personalized or Patient-Specific Insights
WGS platforms go a step further in tailoring insights to individual patients. They prioritize causal genes and affected systems, enabling more personalized therapies. When paired with phenotype-aware AI tools and the Human Phenotype Ontology (HPO), WGS platforms excel at "causal gene discovery." For example, the SHEPHERD deep learning tool correctly identified the causal gene first in 40% of patients and ranked it within the top five for 77.8% of difficult-to-diagnose cases.
"SHEPHERD performs granular, phenotype-based causal gene discovery by ranking candidate genes from bioinformatics pipelines... improving diagnostic efficiency by at least twofold compared to a non-guided baseline."
- Emily Alsentzer et al.
Another advantage of WGS is its ability to provide data that can be reanalyzed as new gene–phenotype relationships are uncovered. This feature is especially beneficial for rare disease patients, many of whom face a diagnostic journey lasting over five years before receiving an accurate diagnosis.
8. Exome Sequencing Tools
Exome sequencing tools zero in on the protein-coding regions of the genome - about 1% of the total DNA - where most mutations linked to diseases are found. These tools play a critical role in diagnosing rare diseases by offering a balance between thorough coverage and cost efficiency. They also use phenotype data to speed up and refine the diagnostic process.
Relevance to Rare Disease Research
When it comes to uncovering disease-causing variants, exome sequencing tools shine, especially when they integrate phenotype data. Take Exomiser, for example - it combines patient symptoms, encoded as Human Phenotype Ontology (HPO) terms, with sequencing data to rank the most likely causative variants. Between February 2019 and February 2022, Genomics England reanalyzed 24,015 unsolved cases from the 100,000 Genomes Project using Exomiser 13.1.0. This effort led to 463 new diagnoses, representing a 2% increase in diagnostic yield. Remarkably, 114 of these diagnoses came from genes that weren’t included in the original disease panels, showcasing the potential of phenotype-driven, panel-independent approaches. This integration of phenotype data is a game-changer for improving diagnostic outcomes.
Accuracy and Reliability of Results
The addition of phenotype data significantly boosts the accuracy of these tools. Studies show that top-rank accuracy jumps from 3% to 74% when phenotype integration is applied. Exomiser, for instance, identifies the correct diagnostic variant as the top-ranked candidate in 71% to 74% of cases. When the top five candidates are considered, sensitivity increases to 92% to 94%.
These tools also use automated ACMG/AMP classification to standardize how variants are categorized. Impressively, this system can reclassify 92% of variants from “uncertain significance” to “pathogenic” or “likely pathogenic”. To further refine results, they filter variants against massive population datasets like gnomAD, 1000 Genomes, TOPMed, and UK10K, typically applying a rarity threshold of less than 0.1% for rare disease candidates.
Support for Personalized or Patient-Specific Insights
Beyond diagnostic accuracy, exome sequencing tools offer insights tailored to a patient’s unique genetic makeup. In October 2025, researchers studied 386 diagnosed probands from the Undiagnosed Diseases Network (UDN). By fine-tuning parameters in Exomiser, they improved the success rate for identifying coding diagnostic variants in the top 10 rankings from 67.3% to 88.2%. The Mosaic platform also played a role in enabling ongoing reanalysis, further increasing the diagnostic yield for previously undiagnosed cases.
With hundreds of new disease-gene associations discovered every year, periodic reinterpretation of exome data using updated databases can boost diagnostic success by 10% to 15%. This capability is especially critical for the 50% to 80% of rare disease patients who remain undiagnosed after their initial sequencing. Additionally, these tools incorporate cross-species data, comparing patient phenotypes to models like mice and zebrafish to prioritize genes that might not yet have known human disease associations.
9. PhenIX
Relevance to Rare Disease Research
PhenIX, short for Phenotypic Interpretation of eXomes, is a computational tool crafted specifically for studying Mendelian disorders. It works by narrowing down genetic variants from exome sequencing or next-generation sequencing (NGS) panels into a concise list of top candidates. This is achieved by linking genetic data with clinical symptoms.
"PhenIX ranks genes based on predicted variant pathogenicity as well as phenotypic similarity of diseases associated with the genes harboring these variants to the phenotypic profile of the individual being investigated, based on analysis powered by the Human Phenotype Ontology (HPO)."
Ability to Integrate Genotype and Phenotype Data
PhenIX excels in connecting genetic and clinical data, making it especially valuable for rare disease research. It combines two critical factors: variant pathogenicity (the likelihood of a genetic change causing disease) and phenotypic similarity (how closely a patient's symptoms align with known diseases). By standardizing patient symptoms using the Human Phenotype Ontology (HPO), PhenIX compares them to thousands of Mendelian diseases cataloged in databases like OMIM and Orphanet. Within the Exomiser framework, users can activate PhenIX by replacing the default setting with "phenixPrioritiser: {}".
Accuracy and Reliability of Results
PhenIX has demonstrated strong performance in identifying causative variants. A study at Moorfields Eye Hospital and the UCL Institute of Ophthalmology tested the tool on whole-exome data from 134 individuals with rare inherited retinal diseases. The results were impressive: PhenIX ranked the correct diagnosis first in 69% (93 out of 134) of cases. When expanding to the top five candidates, the tool successfully identified the causative variants for about 93% of the dataset. The study also noted a mean rank of 2.1 for causative genes, highlighting its consistent accuracy across different rare disease cohorts.
Support for Personalized or Patient-Specific Insights
PhenIX further enhances its utility by offering insights tailored to individual patients. By using a detailed set of HPO terms that describe a patient's clinical symptoms, the tool evaluates the similarity between the patient's profile and known Mendelian diseases. This process generates a personalized ranking of potential causative genes. For the best results, clinicians should provide a comprehensive and precise list of HPO terms to capture the patient's clinical abnormalities accurately.
10. Variant Effect Predictor (VEP)
Relevance to Rare Disease Research
The Ensembl Variant Effect Predictor (VEP) is designed to analyze and prioritize genomic variants across both coding and noncoding regions. With millions of variants present in a typical human genome, only 50–100 are protein-truncating or loss-of-function, making the ability to prioritize these variants essential for researchers. VEP helps by assessing the effects of variants on genes, transcripts, protein sequences, and regulatory regions.
"The Ensembl Variant Effect Predictor is a powerful toolset for the analysis, annotation, and prioritization of genomic variants in coding and non-coding regions."
VEP also bridges genetic findings with clinical databases by leveraging information from resources like OMIM, Orphanet, the GWAS Catalog, and ClinVar. For researchers, the --gene_phenotype flag can pinpoint variants linked to known phenotypes. In March 2026, Genomics England implemented VEP versions 112, 113.3, and 115.2 in its Research Environment. These versions are preloaded with plugins such as CADD (v1.7), SpliceAI, and ClinVar (20240627 release), supporting studies into rare diseases and cancer.
Ability to Integrate Genotype and Phenotype Data
VEP’s plugin system takes variant prioritization a step further by integrating phenotype data. For example, the Phenotypes plugin connects variants to documented human phenotypes, while the G2P (Gene-to-Phenotype) plugin links genes to specific clinical phenotypes like skin disorders. The DisGeNET plugin adds another layer by associating variants with diseases.
VEP also supports haplotype analysis through Haplosaurus, which enables phased genotype analysis to generate observed transcript haplotype sequences. Additionally, it uses Sequence Ontology (SO) terms to describe functional impacts, ensuring clear and consistent communication among researchers.
Accuracy and Reliability of Results
A 2024 systematic review highlighted 118 variant effect predictors, noting that Ensembl VEP is one of only two tools capable of handling seven different variant types. It can predict 54 distinct functional impacts, covering changes in coding sequences, untranslated regions, gene structures, and regulatory elements.
When combined with tools like SnpEff and FAVOR, VEP can predict about 61% (99 out of 161) of all known functional impacts. Plugins such as LOFTEE (for high-confidence loss-of-function variants), SpliceAI (for splicing predictions), and REVEL (for missense pathogenicity) further enhance its reliability.
VEP also integrates allele frequency data from the 1000 Genomes Project and gnomAD, helping researchers differentiate rare, potentially harmful variants from common, benign ones. Impressively, the tool can process a complete human variant set of approximately 4.47 million variants in just 62 minutes on a modern quad-core machine. These capabilities make it a key resource for generating personalized genetic insights.
Support for Personalized or Patient-Specific Insights
VEP allows researchers to tailor analyses, filtering results based on specific gene panels tied to clinical symptoms. The G2P plugin is particularly useful for mapping genes to clinical phenotypes like skin disorders. The OpenTargets plugin adds value by linking variants to potential drug targets, aiding therapeutic development.
For cases where data privacy is critical, VEP offers an offline mode using local cache files, ensuring sensitive clinical data remains secure. It also supports GRCh37 and GRCh38 assemblies, allowing compatibility with a wide range of clinical datasets and older research projects. As an open-source tool, VEP is freely available for academic and commercial use, making it a key player in advancing personalized medicine for rare genetic conditions.
Conclusion
These tools are reshaping the landscape of rare disease research. By integrating standardized clinical vocabularies like the Human Phenotype Ontology with genomic data - such as gnomAD allele frequencies and CADD pathogenicity scores - these platforms take millions of genetic variants and filter them down to a manageable list of likely candidates for deeper investigation. This combination of genotype and phenotype analysis lays the groundwork for precise, patient-focused interventions.
"Knowledge of genotype-phenotype correlations can contribute to our understanding of the pathogenesis, clinical manifestations, prognosis and treatment response for many genetic conditions and cancers."
- Genomics Education Programme
The impact on patient care is undeniable. Tools like DeepRare and SHEPHERD have shown outstanding results in identifying causative variants. For example, deep learning has successfully ranked the causal gene among the top five predictions for 77.8% of patients. This is a significant breakthrough, especially considering that patients with rare diseases often face an average diagnostic journey lasting over five years.
Beyond diagnostics, RareLabs bridges the gap between genetic discoveries and therapeutic solutions. Once causative variants are identified, RareLabs uses patient-derived cell models to explore various treatment options, including small molecule screening, antisense oligonucleotide therapy, and gene replacement therapy. This approach directly connects diagnosis to personalized treatments, offering hope to millions around the globe.
What makes these resources so powerful is how seamlessly they work together. Standardized phenotyping speeds up variant prioritization, statistical models help pinpoint pathogenic variants by predicting inheritance patterns, and discovery platforms uncover new gene-phenotype relationships by clustering unsolved cases with similar clinical traits. Together, these tools not only improve diagnostic precision but also create a direct path from raw data to customized treatments.
FAQs
How do I choose between WGS and exome sequencing?
When deciding between whole genome sequencing (WGS) and exome sequencing (WES), it all comes down to the focus of your analysis.
- WGS offers a comprehensive look at genetic variation, covering both coding and non-coding regions. This makes it especially useful for identifying novel variants, including those outside of known genes.
- WES, on the other hand, zeroes in on exons - the parts of the genome where most disease-causing mutations are found. It's also a more budget-friendly option compared to WGS.
Your decision should align with your specific research objectives, available budget, and whether examining non-coding regions is critical for your study.
Why are HPO terms important for diagnosis?
HPO terms play a key role in diagnosing medical conditions by providing a standardized language to describe phenotypic abnormalities. This standardization ensures that the documentation of symptoms is consistent, which helps in multiple ways:
- Supports clinical decisions: Clear and uniform descriptions make it easier for healthcare providers to assess and manage cases effectively.
- Improves differential diagnosis: With a shared vocabulary, comparing and contrasting potential diagnoses becomes more straightforward.
- Enhances genomic data interpretation: HPO terms bridge the gap between observed symptoms and their potential genetic causes, making it easier to uncover connections.
By using HPO terms, medical professionals can better link a patient’s symptoms to their genetic origins, leading to more accurate and efficient diagnoses.
When should RNA-seq be added to genetic testing?
RNA-seq can be a powerful addition to genetic testing when earlier tests fail to provide clear answers. By analyzing gene expression, RNA-seq offers functional data that can shed light on unresolved rare disease cases. This method goes beyond standard genetic testing, helping to identify hidden genetic causes and offering critical insights into how genes function and contribute to these conditions.



